9 Trabajando con datos de cuentas de experimentos ómicos
Muchos dispositivos de medición en biotecnología se basan en el muestreo y el recuento masivo de moléculas en paralelo. Un ejemplo es la secuenciación de ADN de alto rendimiento. Sus aplicaciones se dividen ampliamente en dos clases principales de salida de datos: en el primer caso, la salida de interés son las secuencias en sí mismas, quizás también sus polimorfismos o diferencias con otras secuencias vistas antes. En el segundo caso, las secuencias en sí son más o menos bien entendidas (digamos, tenemos un genoma bien ensamblado y anotado), y nuestro interés está en cuán abundantes son las diferentes regiones de secuencia en nuestra muestra.
Por ejemplo, en RNA-Seq (Ozsolak y Milos 2011), secuenciamos las moléculas de ARN que se encuentran en una población de células o en un tejido.
Estrictamente hablando, no secuenciamos el ARN sino el ADN complementario (ADNc) obtenido de la transcripción inversa. El conjunto de todo el ARN podría reducirse a un subconjunto de interés (p. Ej., ARN mensajero) por medios bioquímicos, como la selección poli-A o el agotamiento del ARN ribosómico. Existen variantes sensibles de RNA-Seq que permiten analizar células individuales y un gran número de ellas.
En ChIP-Seq, secuenciamos regiones de ADN que están unidas a proteínas de unión a ADN particulares (seleccionadas por inmunoprecipitación); en RIP-Seq, moléculas de ARN o regiones de las mismas unidas a una proteína de unión a ARN particular; en DNA-Seq, secuenciamos el ADN genómico y estamos interesados en la prevalencia de variantes genéticas en poblaciones heterogéneas de células, por ejemplo, la composición clonal de un tumor. En la captura de conformación de cromatina de alto rendimiento (HiC) nuestro objetivo es mapear la disposición espacial 3D del ADN; en screens genéticos (utilizando, digamos, bibliotecas de ARNi o CRISPR-Cas9 para perturbaciones y secuenciación de alto rendimiento para lectura), estamos interesados en la proliferación o supervivencia de células tras la eliminación, desactivación o modificación de genes. En el análisis de microbiomas, estudiamos la abundancia de diferentes especies microbianas en hábitats microbianos complejos.
Idealmente, podríamos querer secuenciar y contar todas las moléculas de interés en la muestra. Generalmente esto no es posible: los protocolos bioquímicos no son 100% eficientes y algunas moléculas o intermedios se pierden en el camino. Además, a menudo ni siquiera es necesario. En su lugar, secuenciamos y contamos una muestra estadística. El tamaño de la muestra dependerá de la complejidad del conjunto de secuencias ensayado; puede pasar de decenas de miles a miles de millones. Esta naturaleza de muestreo de los datos es importante cuando se trata de analizarlos. Esperamos que el muestreo sea lo suficientemente representativo para que podamos identificar tendencias y patrones interesantes.
9.1 Algunos conceptos básicos
Antes de comenzar, establezcamos algunos términos clave.
Una biblioteca de secuenciación es la colección de moléculas de ADN que se utilizan como entrada para la máquina de secuenciación.
Los fragmentos son las moléculas que se secuencian. Dado que la tecnología más utilizada en la actualidad solo puede tratar con moléculas de entre 300 y 1000 nucleótidos de longitud, estos se obtienen fragmentando las moléculas de ADN o ADNc (generalmente más largas) de interés.
Una lectura es la secuencia obtenida de un fragmento. Con la tecnología actual, la lectura no cubre todo el fragmento, sino solo uno o ambos extremos, y la longitud de lectura en cada lado es de alrededor de 150 nucleótidos.
Entre la secuenciación y el recuento, hay un paso importante de agregación o agrupación involucrado, que agrega secuencias que pertenecen juntas: por ejemplo, todas las lecturas que pertenecen al mismo gen (en RNA-Seq), o a la misma región de unión (ChIP-Seq) . Hay varios enfoques para esto y opciones a tomar, dependiendo del objetivo del experimento. Los métodos incluyen alineación explícita o mapeo basado en hash a una secuencia de referencia, y agrupación de lecturas basada en similitud de secuencia independiente de referencia, especialmente si no hay una referencia obvia, como en metagenómica o metatranscriptómica. Necesitamos elegir si considerar diferentes alelos o isoformas por separado, o fusionarlos en una clase de equivalencia. Para simplificar, usaremos el término gen en este capítulo para estos agregados operativos, aunque pueden ser varias cosas dependiendo de la aplicación en particular.
9.2 Contar datos
Carguemos un conjunto de datos de ejemplo. Es parte del paquete de datos del experimento pasilla.
fn = system.file("extdata", "pasilla_gene_counts.tsv",
package = "pasilla", mustWork = TRUE)
counts = as.matrix(read.csv(fn, sep = "\t", row.names = "gene_id"))Los datos se almacenan como una tabla rectangular en un archivo delimitado por tabulaciones, que hemos leído en los recuentos de la matriz.
dim(counts)## [1] 14599 7counts[ 2000+(0:3), ]## untreated1 untreated2 untreated3 untreated4 treated1 treated2
## FBgn0020369 3387 4295 1315 1853 4884 2133
## FBgn0020370 3186 4305 1824 2094 3525 1973
## FBgn0020371 1 0 1 1 1 0
## FBgn0020372 38 84 29 28 63 28
## treated3
## FBgn0020369 2165
## FBgn0020370 2120
## FBgn0020371 0
## FBgn0020372 27La matriz cuenta el número de lecturas observadas para cada gen en cada muestra. Lo llamamos la tabla de conteo. Tiene 14.599 filas, correspondientes a los genes, y 7 columnas, correspondientes a las muestras. Al cargar datos desde un archivo, una buena verificación de plausibilidad es imprimir algunos de los datos, y tal vez no solo al principio, sino también en algún punto aleatorio en el medio, como hicimos anteriormente.
La tabla es una matriz de valores enteros: el valor en la fila i y la columna j de la matriz indica cuántas lecturas se han asignado al gen i en la muestra j. Los modelos de muestreo estadístico que discutimos en este práctico se basan en el hecho de que los valores son los recuentos directos y “brutos” de las lecturas de secuenciación, no una cantidad derivada, como recuentos normalizados, recuentos de pares de bases cubiertos o similares; esto solo conduciría a resultados sin sentido.
9.3 Un análisis básico
Regresemos a los datos de pasilla de la sección. Estos datos provienen de un experimento en cultivos de células de Drosophila melanogaster que investigó el efecto de la eliminación del ARNi del factor de empalme pasilla (Brooks et al. 2011) en el transcriptoma de las células. Hubo dos condiciones experimentales, denominadas no tratadas y tratadas en el encabezado de la tabla de recuento que cargamos. Corresponden al control negativo y al siARN contra pasilla. Los metadatos experimentales de las 7 muestras de este conjunto de datos se proporcionan en una tabla similar a una hoja de cálculo, que cargamos.
annotationFile = system.file("extdata",
"pasilla_sample_annotation.csv",
package = "pasilla", mustWork = TRUE)
pasillaSampleAnno = readr::read_csv(annotationFile)##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## file = col_character(),
## condition = col_character(),
## type = col_character(),
## `number of lanes` = col_double(),
## `total number of reads` = col_character(),
## `exon counts` = col_double()
## )pasillaSampleAnno## # A tibble: 7 x 6
## file condition type `number of lane… `total number of r… `exon counts`
## <chr> <chr> <chr> <dbl> <chr> <dbl>
## 1 treated… treated single-… 5 35158667 15679615
## 2 treated… treated paired-… 2 12242535 (x2) 15620018
## 3 treated… treated paired-… 2 12443664 (x2) 12733865
## 4 untreat… untreated single-… 2 17812866 14924838
## 5 untreat… untreated single-… 6 34284521 20764558
## 6 untreat… untreated paired-… 2 10542625 (x2) 10283129
## 7 untreat… untreated paired-… 2 12214974 (x2) 11653031Como vemos aquí, el conjunto de datos general se produjo en dos lotes, el primero que consta de tres bibliotecas de secuenciación que se sometieron a secuenciación de lectura única, el segundo lote que consta de cuatro bibliotecas para las que se utilizó la secuenciación de extremos emparejados. Como sucede a menudo, necesitamos hacer algunos cambios de datos: reemplazamos los guiones en la columna de tipo por guiones bajos, ya que DESeq2 desaconseja los operadores aritméticos en niveles de factor, y convertimos las columnas de tipo y condición en factores, especificando explícitamente nuestro orden preferido de los niveles (el valor predeterminado es alfabético).
library("dplyr")
pasillaSampleAnno = mutate(pasillaSampleAnno,
condition = factor(condition, levels = c("untreated", "treated")),
type = factor(sub("-.*", "", type), levels = c("single", "paired")))Observamos que el diseño está aproximadamente equilibrado entre el factor de interés, la condición y el tipo de “factor de molestia”:
with(pasillaSampleAnno,
table(condition, type))## type
## condition single paired
## untreated 2 2
## treated 1 2DESeq2 usa un contenedor de datos especializado, llamado DESeqDataSet para almacenar los conjuntos de datos con los que trabaja. Tal uso de contenedores especializados - o, en terminología R, clases - es un principio común del proyecto Bioconductor, ya que ayuda a los usuarios a mantener juntos los datos relacionados. Si bien esta forma de hacer las cosas requiere que los usuarios inviertan un poco más de tiempo por adelantado para comprender las clases, en comparación con solo usar tipos de datos R básicos como matriz y dataframe, ayuda a evitar errores debido a la pérdida de sincronización entre partes relacionadas de los datos. También permite la abstracción y encapsulación de operaciones comunes que podrían ser bastante prolijas si siempre se expresan en términos básicos. DESeqDataSet es una extensión de la clase SummarizedExperiment en Bioconductor. La clase SummarizedExperiment también es utilizada por muchos otros paquetes, por lo que aprender a trabajar con ella le permitirá utilizar una gran variedad de herramientas.
Usamos la función constructora DESeqDataSetFromMatrix para crear un DESeqDataSet a partir de los recuentos de la matriz de datos de recuento y el dataframe de anotación de muestra pasillaSampleAnno.
mt = match(colnames(counts), sub("fb$", "", pasillaSampleAnno$file))
stopifnot(!any(is.na(mt)))
library("DESeq2")
pasilla = DESeqDataSetFromMatrix(
countData = counts,
colData = pasillaSampleAnno[mt, ],
design = ~ condition)
class(pasilla)## [1] "DESeqDataSet"
## attr(,"package")
## [1] "DESeq2"Tenga en cuenta que en el siguiente código, tenemos que hacer un trabajo adicional para hacer coincidir los nombres de columna del objeto count con la columna de archivo del dataframe pasillaSampleAnno, en particular, debemos eliminar el “fb” que se usa en el columna de archivo por alguna razón. Esta disputa de datos es muy común. Una de las razones para almacenar los datos en un objeto DESeqDataSet es que entonces ya no tenemos que preocuparnos por esas cosas.
is(pasilla, "SummarizedExperiment")## [1] TRUELa clase SummarizedExperiment, y por lo tanto DESeqDataSet, también contiene funciones para almacenar anotaciones de las filas de la matriz de conteo. Por ahora, estamos contentos con los identificadores de genes de los nombres de fila de la tabla de conteos.
El método DESeq2
Después de estos preparativos, ahora estamos listos para saltar directamente al análisis de expresión diferencial. Nuestro objetivo es identificar genes que son diferencialmente abundantes entre las células tratadas y no tratadas. Con este fin, aplicaremos una prueba que es conceptualmente similar a la prueba t, aunque matemáticamente algo más compleja. Una selección de pasos de análisis estándar se incluye en una sola función, DESeq.
pasilla = DESeq(pasilla)## estimating size factors## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testingLa función DESeq es simplemente un contenedor que llama, en orden, a las funciones estimateSizeFactors, estimateDispersions (estimación de dispersión) y nbinomWaldTest (pruebas de hipótesis para la abundancia diferencial). La prueba se encuentra entre los dos niveles textttuntreated y texttttreated del factor condition, ya que esto es lo que especificamos cuando construimos el objeto pasilla mediante el argumento design = simcondition. Siempre puede llamar a cada una de estas tres funciones individualmente si desea modificar su comportamiento o introducir pasos personalizados. Miremos results.
res = results(pasilla)
res[order(res$padj), ] %>% head## log2 fold change (MLE): condition treated vs untreated
## Wald test p-value: condition treated vs untreated
## DataFrame with 6 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## FBgn0039155 730.596 -4.61901 0.1687068 -27.3789 4.88599e-165
## FBgn0025111 1501.411 2.89986 0.1269205 22.8479 1.53430e-115
## FBgn0029167 3706.117 -2.19700 0.0969888 -22.6521 1.33042e-113
## FBgn0003360 4343.035 -3.17967 0.1435264 -22.1539 9.56283e-109
## FBgn0035085 638.233 -2.56041 0.1372952 -18.6490 1.28772e-77
## FBgn0039827 261.916 -4.16252 0.2325888 -17.8965 1.25663e-71
## padj
## <numeric>
## FBgn0039155 4.06661e-161
## FBgn0025111 6.38497e-112
## FBgn0029167 3.69104e-110
## FBgn0003360 1.98979e-105
## FBgn0035085 2.14354e-74
## FBgn0039827 1.74316e-68Explorando los resultados
El primer paso después de un análisis de expresión diferencial es la visualización de las siguientes tres o cuatro gráficos básicos:
el histograma de valores p,
la gráfica MA y
un diagrama de ordenación.
Además, un mapa de calor puede resultar útil.
Estas son medidas esenciales de evaluación de la calidad de los datos, y el consejo general sobre evaluación y control de la calidad.
El histograma del p-value es sencillo.
ggplot(as(res, "data.frame"), aes(x = pvalue)) +
geom_histogram(binwidth = 0.01, fill = "Royalblue", boundary = 0)## Warning: Removed 2241 rows containing non-finite values (stat_bin).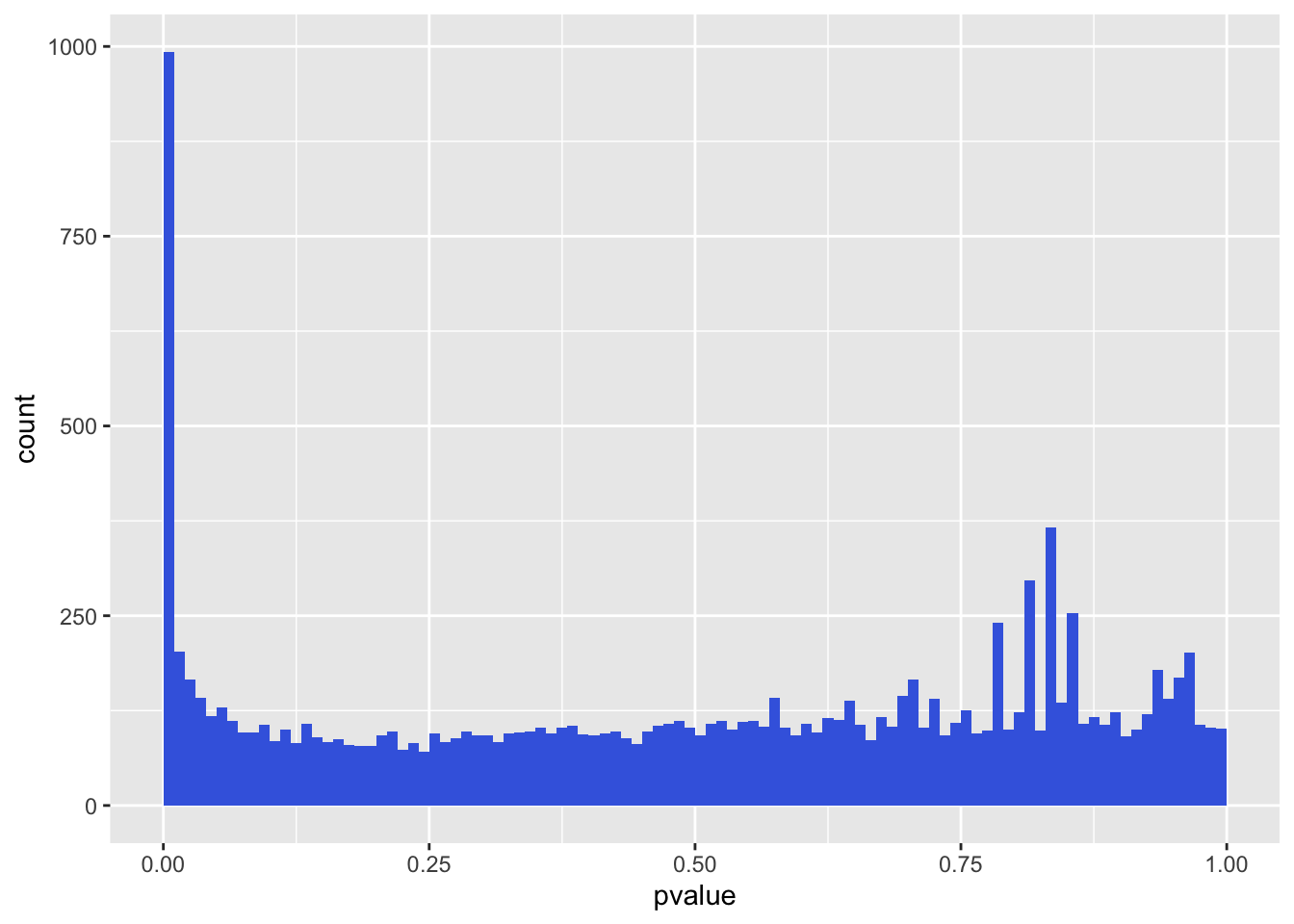
Histograma de valores p de un análisis de expresión diferencial.
La distribución muestra dos componentes principales: un fondo uniforme con valores entre 0 y 1, y un pico de valores p pequeños a la izquierda. El fondo uniforme corresponde a los genes no expresados diferencialmente. Por lo general, esta es la mayoría de los genes. El pico de la izquierda corresponde a genes expresados diferencialmente. Como ya vimos, la relación entre el nivel del fondo y la altura del pico nos da una indicación aproximada de la tasa de descubrimiento falso (FDR) que estaría asociada con llamar a los genes en el grupo más a la izquierda expresados diferencialmente. En nuestro caso, el grupo más a la izquierda contiene todos los valores de p entre 0 y 0,01, que corresponden a 993 genes. El nivel de fondo es de alrededor de 100, por lo que el FDR asociado con llamar a todos los genes en el contenedor más a la izquierda sería de alrededor del 10%.
Para producir el gráfico MA, podemos usar la función plotMA en el paquete DESeq2
plotMA(pasilla, ylim = c( -2, 2))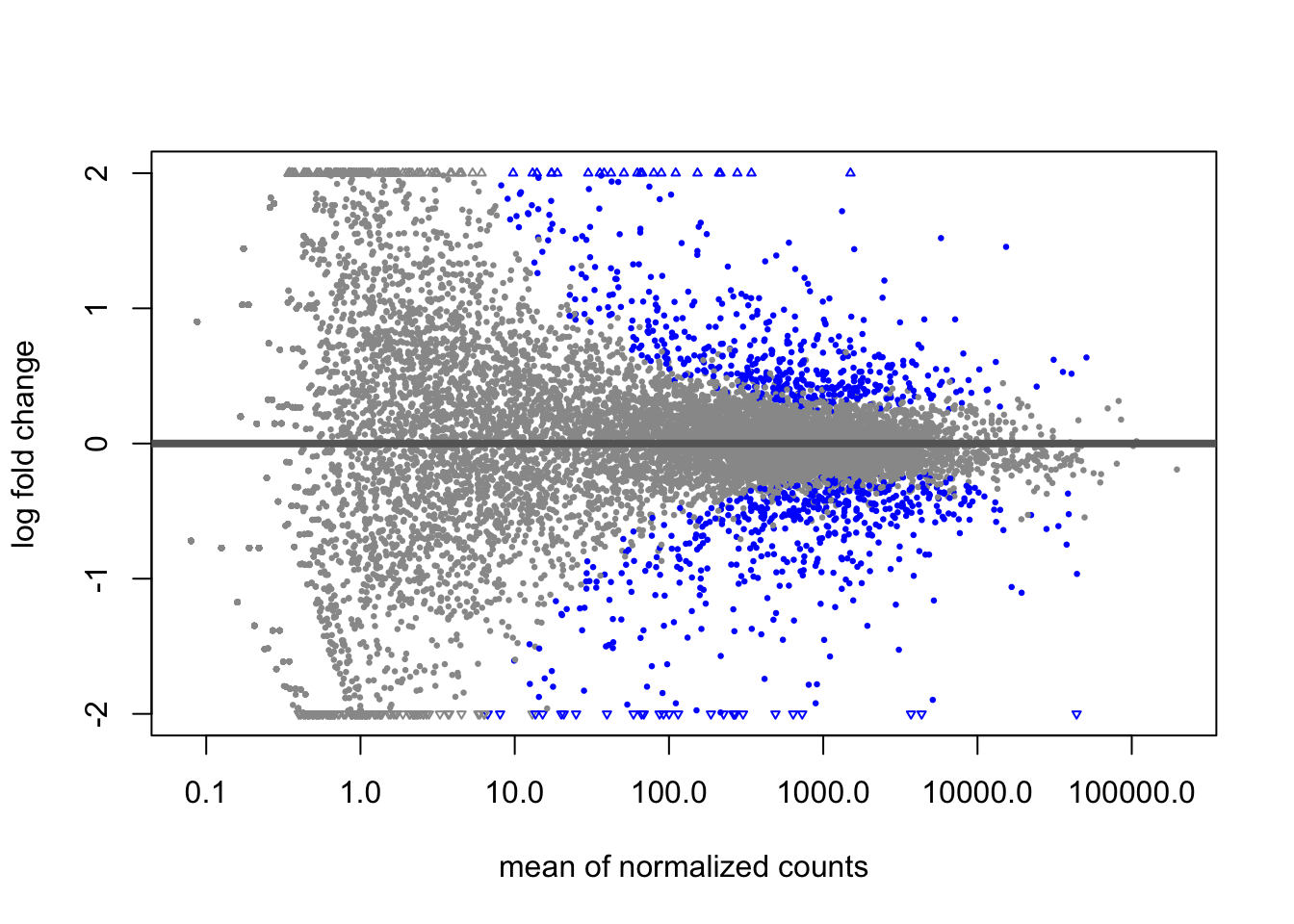
Ceces de cambio frente a la media de los recuentos normalizados por factor de tamaño. La escala logarítmica se utiliza para ambos ejes. De forma predeterminada, los puntos se colorean en rojo si el valor p ajustado es inferior a 0,1. Los puntos que caen fuera del rango del eje y se representan como triángulos.
Para producir gráficos de PCA similares a los que vimos en el práctico de Análisis Multivariado, podemos usar la función DESeq2 plotPCA
pas_rlog = rlogTransformation(pasilla)
plotPCA(pas_rlog, intgroup=c("condition", "type")) + coord_fixed()## Coordinate system already present. Adding new coordinate system, which will replace the existing one.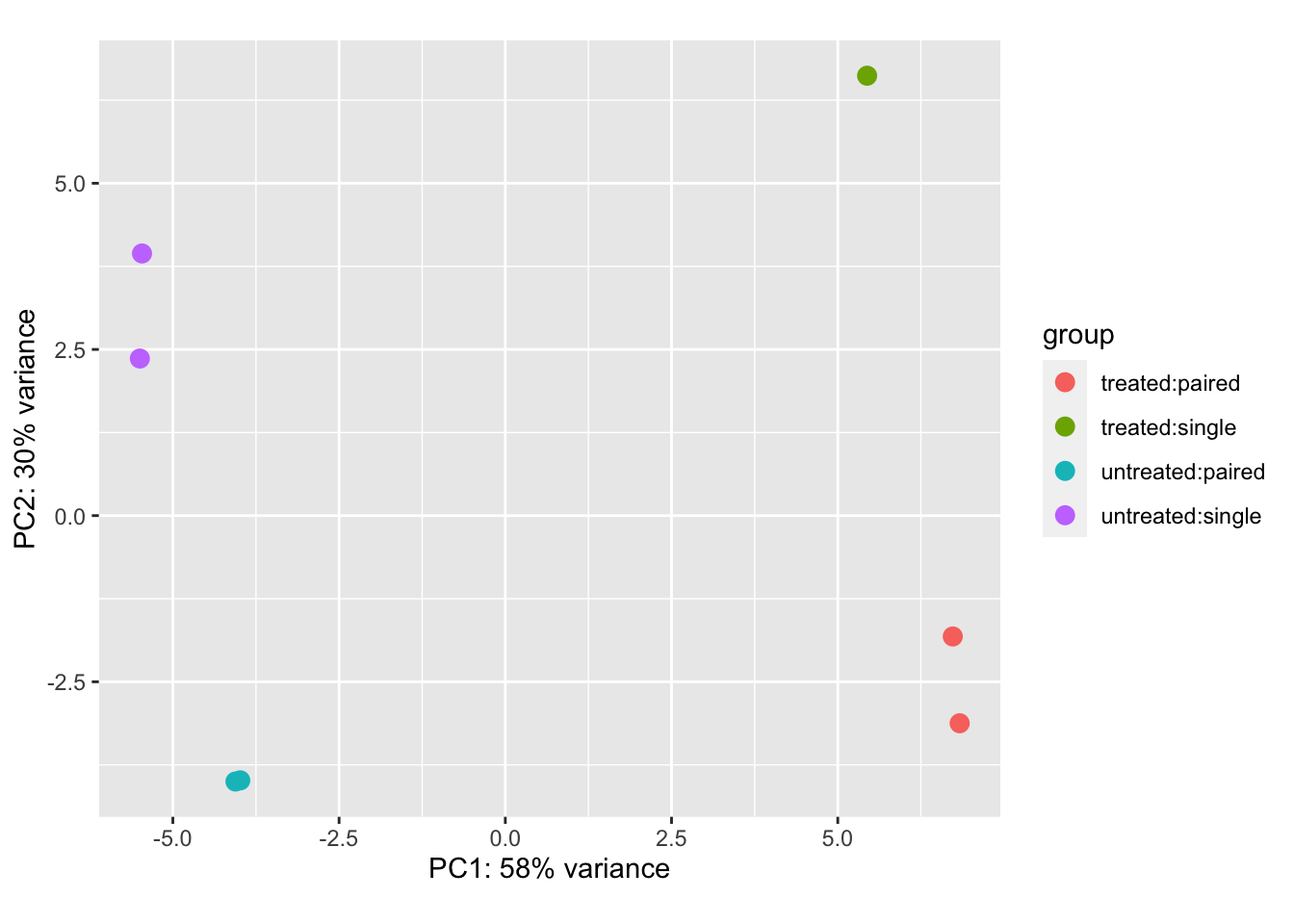
Como vimos en el capítulo anterior, este tipo de gráfico es útil para visualizar el efecto general de las covariables experimentales y / o para detectar efectos por lotes. Aquí, el primer eje principal, PC1, está mayormente alineado con la covariable experimental de interés (no tratado /tratado), mientras que el segundo eje está alineado aproximadamente con el protocolo de secuenciación (single/paired).
Exportando los resultados
Se puede exportar un reporte HTML de los resultados con gráficos y columnas clasificables / filtrables utilizando el paquete ReportingTools en un DESeqDataSet que ha sido procesado por la función DESeq. Para ver un ejemplo de código, consulte la viñeta de expresión diferencial RNA-Seq del paquete ReportingTools o la página del manual del método de publicación para la clase DESeqDataSet.
Se puede exportar un archivo CSV de los resultados usando write.csv (o su contraparte del paquete readr).
write.csv(as.data.frame(res), file = "treated_vs_untreated.csv")
9.4 Análisis de dos factores de los datos de pasilla
Además del tratamiento con siARN, que ya hemos considerado en la sección de análisis básicos de datos, los datos de pasilla tienen otra covariable, tipo, que indica el tipo de secuenciación que se realizó.
Vimos en los gráficos de análisis exploratorio de datos (EDA) en que este último tuvo un efecto sistemático considerable en los datos. Nuestro análisis básico no tuvo esto en cuenta, pero lo haremos ahora. Esto debería ayudarnos a obtener una imagen más correcta de qué diferencias en los datos son atribuibles al tratamiento y cuáles se confunden (o se enmascaran) por el tipo de secuenciación.
pasillaTwoFactor = pasilla
design(pasillaTwoFactor) = formula(~ type + condition)
pasillaTwoFactor = DESeq(pasillaTwoFactor)## using pre-existing size factors## estimating dispersions## found already estimated dispersions, replacing these## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testingDe las dos variables tipo y condición, la de interés principal es condición, y en DESeq2, la convención es ponerla al final de la fórmula. Esta convención no tiene ningún efecto sobre el ajuste del modelo, pero ayuda a simplificar algunos de los informes de resultados posteriores. Nuevamente, accedemos a los resultados utilizando la función results, que devuelve un dataframe con las estadísticas de cada gen.
res2 = results(pasillaTwoFactor)
head(res2, n = 3)## log2 fold change (MLE): condition treated vs untreated
## Wald test p-value: condition treated vs untreated
## DataFrame with 3 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## FBgn0000003 0.171569 0.6745518 3.871091 0.1742537 0.861666 NA
## FBgn0000008 95.144079 -0.0406731 0.222215 -0.1830351 0.854770 0.951975
## FBgn0000014 1.056572 -0.0849880 2.111821 -0.0402439 0.967899 NATambién es posible recuperar las veces de cambio de log2, los p-value y los p-value ajustados asociados con la variable de tipo. Los resultados de la función toman un contraste de argumento que permite a los usuarios especificar el nombre de la variable, el nivel que corresponde al numerador de las veces de cambio y el nivel que corresponde al denominador de las veces de cambio (fold change).
resType = results(pasillaTwoFactor,
contrast = c("type", "single", "paired"))
head(resType, n = 3)## log2 fold change (MLE): type single vs paired
## Wald test p-value: type single vs paired
## DataFrame with 3 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## FBgn0000003 0.171569 -1.611546 3.871083 -0.416304 0.677188 NA
## FBgn0000008 95.144079 -0.262255 0.220686 -1.188362 0.234691 0.543822
## FBgn0000014 1.056572 3.290586 2.087243 1.576522 0.114905 NAEntonces, ¿qué obtuvimos de este análisis que tuvo en cuenta el tipo como un factor de molestia (a veces también llamado, más cortésmente, un factor de bloqueo), en comparación con la simple comparación entre dos grupos de la sección de análisis básicos? Grafiquemos los p-value de ambos análisis uno frente al otro.
trsf = function(x) ifelse(is.na(x), 0, (-log10(x)) ^ (1/6))
ggplot(tibble(pOne = res$pvalue,
pTwo = res2$pvalue),
aes(x = trsf(pOne), y = trsf(pTwo))) +
geom_hex(bins = 75) + coord_fixed() +
xlab("Single factor analysis (condition)") +
ylab("Two factor analysis (type + condition)") +
geom_abline(col = "orange")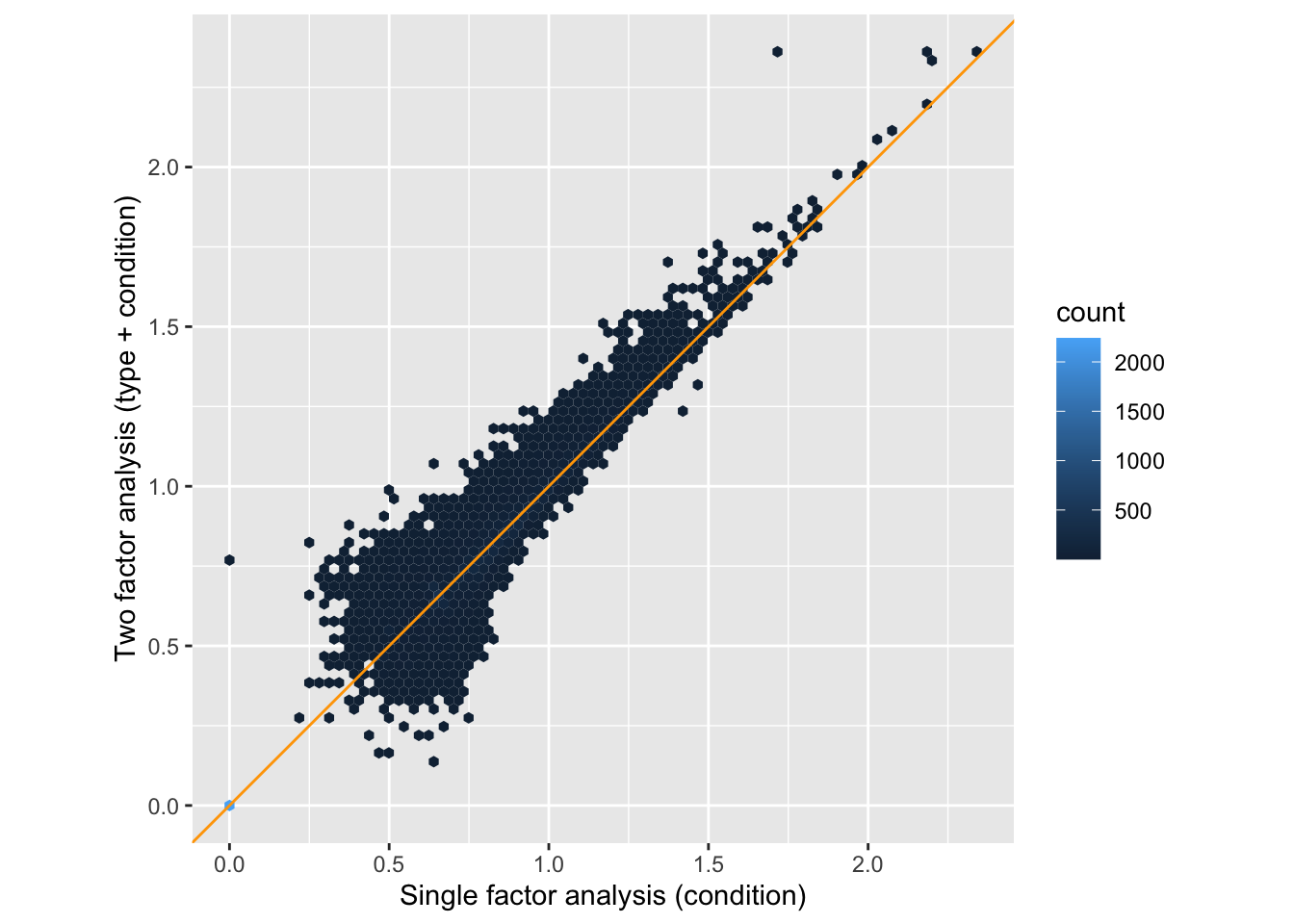
Comparación de los p-value de los modelos con un solo factor (condición) y con dos factores (tipo + condición). Los ejes corresponden a \(log_{10}p^{1/6}\), una transformación monótona decreciente elegida arbitrariamente que comprime el rango dinámico de los valores p con fines de visualización. Podemos ver una tendencia de la distribución conjunta a situarse por encima de la bisectriz, lo que indica que los valores p pequeños en el análisis de dos factores son generalmente más pequeños que los del análisis de un factor.
Como podemos ver en la figura, los valores p en el análisis de dos factores son similares a los del análisis de un factor, pero generalmente son más pequeños. El análisis más sofisticado ha dado lugar a un aumento de poder, aunque modesto. También podemos ver esto contando el número de genes que pasan un cierto umbral de importancia en cada caso:
compareRes = table(
`simple analysis` = res$padj < 0.1,
`two factor` = res2$padj < 0.1 )
addmargins( compareRes )## two factor
## simple analysis FALSE TRUE Sum
## FALSE 6973 289 7262
## TRUE 25 1036 1061
## Sum 6998 1325 8323El análisis de dos factores encontró 1325 genes expresados diferencialmente en un umbral de FDR del 10%, mientras que el análisis de un factor encontró 1061. El análisis de dos factores ha aumentado el poder de detección. En general, la ganancia puede ser incluso mucho mayor, o también menor, dependiendo de los datos. La elección adecuada del modelo requiere una adaptación informada al diseño experimental y la calidad de los datos.