8 Análisis multivariado
En este práctico trabajamos en base al capítulo 7 del libro Moderd Statics for Modern Biology . Pueden revisar y practicar con los ejercicios propuestos en el libro.
Muchos conjuntos de datos constan de varias variables medidas en el mismo conjunto de sujetos: pacientes, muestras u organismos. Por ejemplo, podemos tener características biométricas como altura, peso, edad, así como variables clínicas como presión arterial, azúcar en sangre, frecuencia cardíaca y datos genéticos para, digamos, mil pacientes. La razón de ser del análisis multivariado es la investigación de conexiones o asociaciones entre las diferentes variables medidas. Por lo general, los datos se informan en una estructura de datos tabular con una fila para cada tema y una columna para cada variable. A continuación, nos centraremos en el caso especial en el que cada una de las variables es numérica, por lo que podemos representar la estructura de datos como una matriz en R.
Si las columnas de la matriz son todas independientes entre sí (no relacionadas), podemos simplemente estudiar cada columna por separado y hacer estadísticas estándar “univariadas” sobre ellas una por una; no habría ningún beneficio en estudiarlos como una matriz.
Más a menudo, habrá patrones y dependencias. Por ejemplo, en la biología de las células, sabemos que la tasa de proliferación influirá en la expresión de muchos genes simultáneamente. Al estudiar la expresión de 25.000 genes (columnas) en muchas muestras (filas) de células derivadas de pacientes, notamos que muchos de los genes actúan juntos, ya sea que estén correlacionados positivamente o que estén anti-correlacionados. Perderíamos mucha información importante si solo estudiáramos cada gen por separado. Las conexiones importantes entre genes son detectables solo si consideramos los datos como un todo: cada fila representa las muchas mediciones realizadas en la misma unidad de observación. Sin embargo, tener 25.000 dimensiones de variación para considerar a la vez es abrumador; le mostraremos cómo reducir nuestros datos a un número menor de las dimensiones más importantes sin perder demasiada información.
Este práctico presenta muchos ejemplos de matrices de datos multivariantes que encontramos en experimentos de alto rendimiento, así como algunos ejemplos más elementales que esperamos mejoren su intuición. Nos centraremos en el análisis de componentes principales, abreviado como PCA, un método de reducción de dimensiones. Proporcionaremos explicaciones geométricas del algoritmo, así como visualizaciones que ayudarán a interpretar el resultado de los análisis de PCA.
Para trabajar en estos 3 capitulos es necesario instalar paquetes contenidos en el siguiente enlace. Basta con correr la siguiente línea de comando":
8.1 ¿Cuáles son los datos? Matrices y su motivación
Primero, veamos un conjunto de ejemplos de matrices rectangulares que se usan para representar tablas de medidas. En cada matriz, las filas y columnas representan entidades específicas. Tortugas: Un conjunto de datos muy simple que nos ayudará a comprender los principios básicos es una matriz de tres dimensiones de medidas biométricas en tortugas pintadas.
turtles = read.table(file = "data_cap8910/PaintedTurtles.txt", header = TRUE)
turtles[1:4, ]## sex length width height
## 1 f 98 81 38
## 2 f 103 84 38
## 3 f 103 86 42
## 4 f 105 86 40Las últimas tres columnas son medidas de longitud (en milímetros), mientras que la primera columna es un factor variable que nos dice el sexo de cada animal.
Atletas: esta matriz es un ejemplo interesante del mundo del deporte. Informa las actuaciones de 33 atletas en las diez disciplinas del decatlón: m100, m400 y m1500 son tiempos en segundos para los 100 metros, 400 metros y 1500 metros respectivamente; m110 es el momento de finalizar los 110 metros vallas; la pértiga es la altura del salto con pértiga, y la altura y la longitud son el resultado de los saltos altos y largos, todos en metros; peso, disco y jabalina son las longitudes en metros que los atletas pudieron lanzar el peso, el disco y la jabalina. Aquí están estas variables para los primeros tres atletas:
load("data_cap8910/athletes.RData")
athletes[1:3, ]## m100 long weight highj m400 m110 disc pole javel m1500
## 1 11.25 7.43 15.48 2.27 48.90 15.13 49.28 4.7 61.32 268.95
## 2 10.87 7.45 14.97 1.97 47.71 14.46 44.36 5.1 61.76 273.02
## 3 11.18 7.44 14.20 1.97 48.29 14.81 43.66 5.2 64.16 263.20Tipos de células: Holmes et al. (2005) estudiaron los perfiles de expresión génica de poblaciones de células T clasificadas de diferentes sujetos. Las columnas son un subconjunto de medidas de expresión génica, corresponden a 156 genes que muestran expresión diferencial entre tipos de células.
load("data_cap8910/Msig3transp.RData")
round(Msig3transp,2)[1:5, 1:6]## X3968 X14831 X13492 X5108 X16348 X585
## HEA26_EFFE_1 -2.61 -1.19 -0.06 -0.15 0.52 -0.02
## HEA26_MEM_1 -2.26 -0.47 0.28 0.54 -0.37 0.11
## HEA26_NAI_1 -0.27 0.82 0.81 0.72 -0.90 0.75
## MEL36_EFFE_1 -2.24 -1.08 -0.24 -0.18 0.64 0.01
## MEL36_MEM_1 -2.68 -0.15 0.25 0.95 -0.20 0.17Abundancia de especies bacterianas: las matrices de recuentos se utilizan en estudios de ecología microbiana. Aquí las columnas representan diferentes especies (o unidades taxonómicas operativas, OTU) de bacterias, que se identifican mediante etiquetas numéricas. Las filas están etiquetadas según las muestras en las que se midieron, y los números (enteros) representan el número de veces que se observó cada una de las OTU en cada una de las muestras.
data("GlobalPatterns", package = "phyloseq")
GPOTUs = as.matrix(t(phyloseq::otu_table(GlobalPatterns)))
GPOTUs[1:4, 6:13]## OTU Table: [8 taxa and 4 samples]
## taxa are columns
## 246140 143239 244960 255340 144887 141782 215972 31759
## CL3 0 7 0 153 3 9 0 0
## CC1 0 1 0 194 5 35 3 1
## SV1 0 0 0 0 0 0 0 0
## M31Fcsw 0 0 0 0 0 0 0 0Lecturas de ARNm: Los datos de transcriptoma de RNA-Seq informan el número de lecturas de secuencia que coinciden con cada gen en cada una de varias muestras biológicas.
library("SummarizedExperiment")
data("airway", package = "airway")
assay(airway)[1:3, 1:4]## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## ENSG00000000003 679 448 873 408
## ENSG00000000005 0 0 0 0
## ENSG00000000419 467 515 621 365Es habitual en el campo RNA-Seq, y lo mismo ocurre con los datos de las vías respiratorias anteriores, informar los genes en las filas y las muestras en las columnas. En comparación con las otras matrices que vemos aquí, esto se transpone: las filas y las columnas se intercambian. Convenciones tan diferentes conducen fácilmente a errores, por lo que vale la pena prestarles atención. Perfiles proteómicos: aquí, las columnas son picos de espectroscopía de masas alineados o moléculas identificadas a través de sus relaciones m / z; las entradas en la matriz son las intensidades medidas.
8.2 Preparación y resúmenes de datos de baja dimensión

Si estamos estudiando solo una variable, es decir, solo la tercera columna de la matriz de tortugas, decimos que estamos viendo datos unidimensionales. Dicho vector, digamos todos los pesos de las tortugas, se puede visualizar mediante gráficos como un histograma. Si calculamos un resumen de un número, digamos media o mediana, habremos hecho un resumen de dimensión cero de nuestros datos unidimensionales. Este ya es un ejemplo de reducción de dimensiones.
Si hay demasiadas observaciones, puede ser beneficioso agrupar los datos en contenedores (hexagonales): estos son histogramas bidimensionales. Cuando se consideran dos variables (x e y) medidas juntas en un conjunto de observaciones, el coeficiente de correlación mide cómo las variables covarían. Este es un resumen de un solo número de datos bidimensionales. Su fórmula incluye los resúmenes ¯x y ¯y:
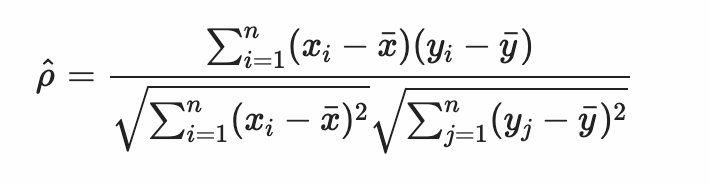
En R, usamos la función cor para calcular su valor. Aplicada a una matriz, esta función calcula todas las correlaciones bidireccionales entre variables continuas.
Calcule la matriz de todas las correlaciones entre las medidas de los datos de las tortugas.¿Qué notaste?
Sacamos la variable categórica y calculamos la matriz.
cor(turtles[, -1])## length width height
## length 1.0000000 0.9783116 0.9646946
## width 0.9783116 1.0000000 0.9605705
## height 0.9646946 0.9605705 1.0000000Vemos que esta matriz cuadrada es simétrica y todos los valores están cerca de 1. Los valores diagonales son siempre 1.
Siempre es beneficioso comenzar un análisis multidimensional verificando estas estadísticas de resumen unidimensionales y bidimensionales simples y hacer presentaciones visuales.
Haz una gráfica de pares de los datos de los atletas. ¿Que notaste?
library("pheatmap")
pheatmap(cor(athletes), cell.width = 10, cell.height = 10)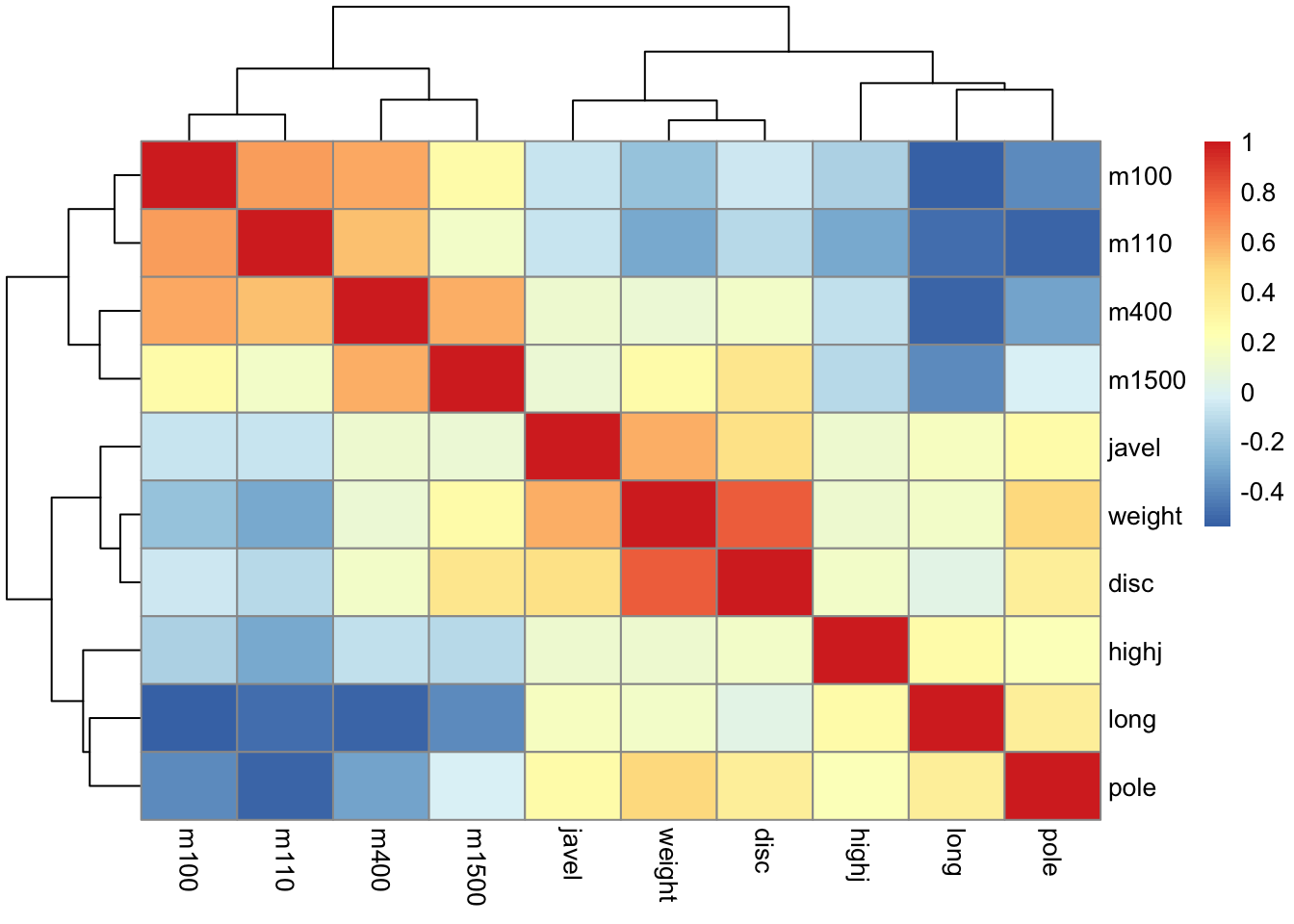 Heatmap de correlaciones entre variables en los datos de los deportistas. Los valores más altos están codificados por colores rojo-naranja. La agrupación jerárquica muestra la agrupación de las disciplinas relacionadas.
Heatmap de correlaciones entre variables en los datos de los deportistas. Los valores más altos están codificados por colores rojo-naranja. La agrupación jerárquica muestra la agrupación de las disciplinas relacionadas.
La figura muestra cómo la agrupación jerárquica de las variables las agrupa en tres grupos: correr, lanzar y saltar.
8.3 Pre procesar los datos
En muchos casos, las diferentes variables se miden en diferentes unidades, por lo que tienen diferentes líneas de base y diferentes escalas, que no son directamente comparables en su forma original.
Para PCA y muchos otros métodos, por lo tanto, necesitamos transformar los valores numéricos a una escala común para que las comparaciones sean significativas. Centrar significa restar la media, de modo que la media de los datos centrados esté en el origen. Escalar o estandarizar significa dividir por la desviación estándar, de modo que la nueva desviación estándar sea 1. De hecho, ya nos hemos encontrado con estas operaciones al calcular el coeficiente de correlación (ecuación previamente mostrada): el coeficiente de correlación es simplemente el producto vectorial de las variables centradas y escaladas. Para realizar estas operaciones, existe la escala de función R, cuyo comportamiento por defecto cuando se le da una matriz o un dataframe es hacer que cada columna tenga una media de cero y una desviación estándar de 1.
apply(turtles[,-1], 2, sd)## length width height
## 20.481602 12.675838 8.392837apply(turtles[,-1], 2, mean)## length width height
## 124.68750 95.43750 46.33333scaledTurtles = scale(turtles[, -1])
apply(scaledTurtles, 2, mean)## length width height
## -1.432050e-18 1.940383e-17 -2.870967e-16apply(scaledTurtles, 2, sd)## length width height
## 1 1 1library(tidyverse)
library(dplyr)
data.frame(scaledTurtles, sex = turtles[, 1]) %>%
ggplot(aes(x = width, y = height, group = sex)) +
geom_point(aes(color = sex)) + coord_fixed()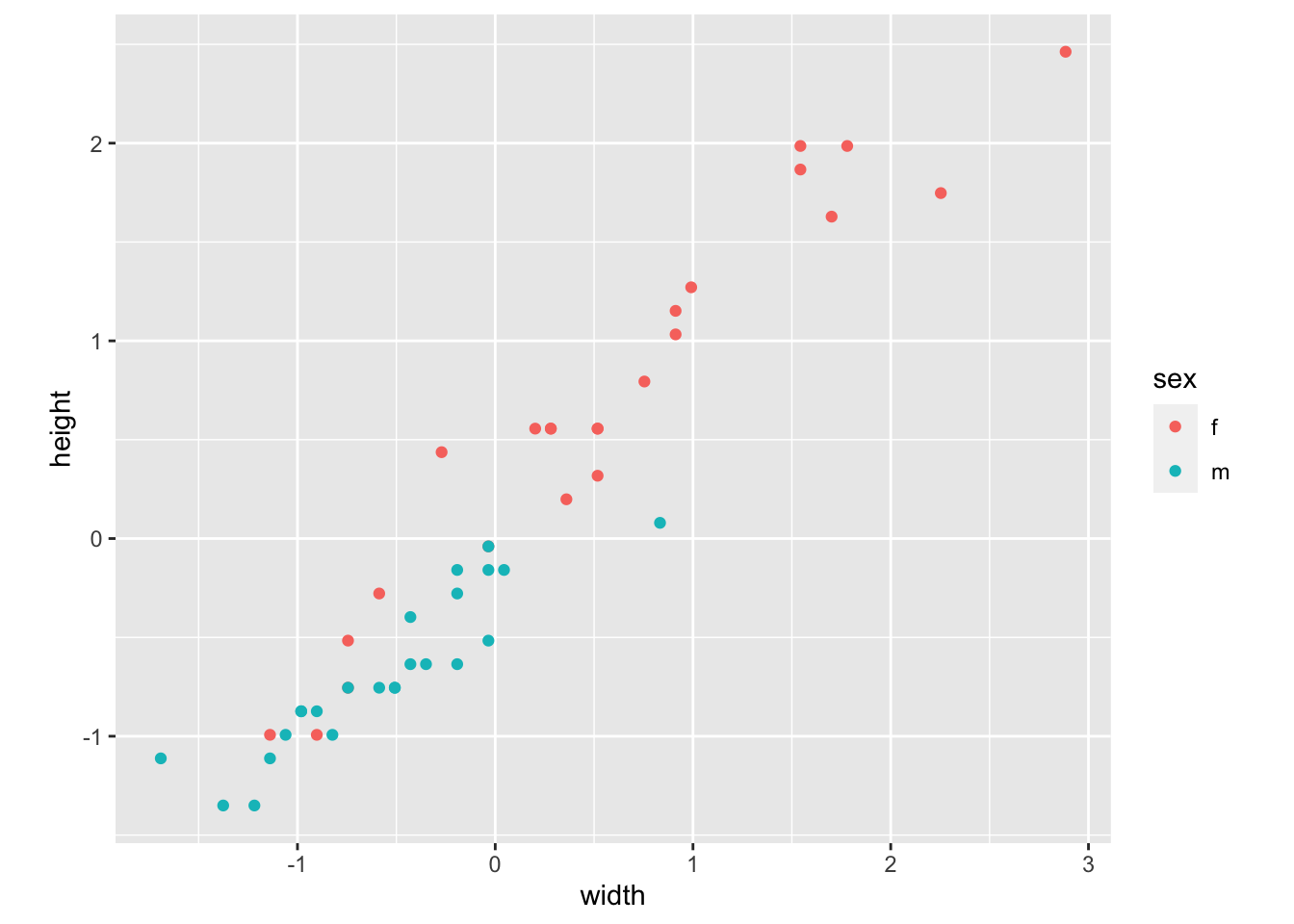
Datos de tortugas proyectados en el plano definido por las variables ancho y alto: cada punto coloreado según el sexo.
Después de preprocesar los datos, estamos listos para emprender la simplificación de datos mediante la reducción de dimensiones.
8.4 Reducción de dimensión
Explicaremos la reducción de dimensiones desde varias perspectivas diferentes. Fue inventado en 1901 por Karl Pearson (Pearson 1901) como una forma de reducir un diagrama de dispersión de dos variables a una sola coordenada. Fue utilizado por estadísticos en la década de 1930 para resumir una batería de pruebas psicológicas ejecutadas en los mismos sujetos (Hotelling 1933); proporcionando así puntuaciones generales que resumen muchas variables de prueba a la vez. Esta idea de puntajes principales inspiró el nombre Análisis de componentes principales (abreviado PCA). El PCA se denomina técnica de aprendizaje no supervisado porque, al igual que en la agrupación, trata todas las variables como si tuvieran el mismo estado. No estamos tratando de predecir o explicar el valor de una variable en particular de las demás; más bien, estamos tratando de encontrar un modelo matemático para una estructura subyacente para todas las variables. PCA es principalmente una técnica exploratoria que produce mapas que muestran las relaciones entre variables y entre observaciones de una manera útil.
PCA es una técnica lineal, lo que significa que buscamos relaciones lineales entre variables y que usaremos nuevas variables que son funciones lineales de las originales.
Las restricciones de linealidad hacen que los cálculos sean particularmente fáciles.
Proyecciones de menor dimensión
Aquí mostramos una forma de proyectar datos bidimensionales en una línea utilizando los datos de los atletas.
athletes = data.frame(scale(athletes))
ath_gg = ggplot(athletes, aes(x = weight, y = disc)) +
geom_point(size = 2, shape = 21)
ath_gg + geom_point(aes(y = 0), colour = "red") +
geom_segment(aes(xend = weight, yend = 0), linetype = "dashed")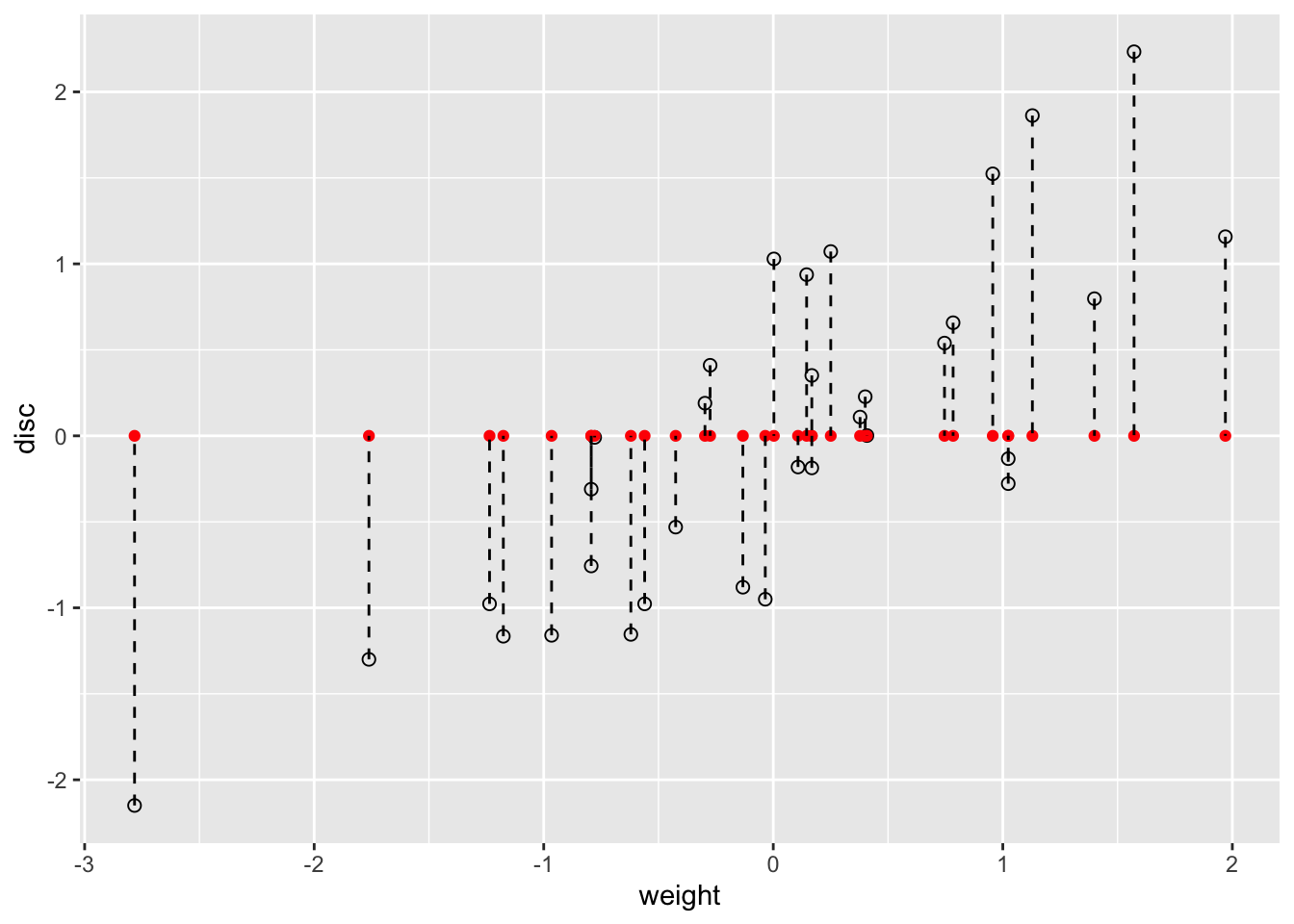 Diagrama de dispersión de dos variables que muestra la proyección en el eje x horizontal (definido por y = 0) en rojo y las líneas de proyección aparecen como discontinuas.
Diagrama de dispersión de dos variables que muestra la proyección en el eje x horizontal (definido por y = 0) en rojo y las líneas de proyección aparecen como discontinuas.
¿Cómo resumimos los datos bidimensionales mediante una línea?
En general, perdemos información sobre los puntos cuando proyectamos desde dos dimensiones (un plano) a una (una línea). Si lo hacemos simplemente usando las coordenadas originales, como hicimos con la variable de peso en la figura recién generada, perdemos toda la información sobre la variable de disco. Nuestro objetivo es mantener la mayor cantidad de información posible sobre ambas variables. En realidad, hay muchas formas de proyectar la nube de puntos en una línea. Uno es usar lo que se conoce como líneas de regresión. Veamos estas líneas y cómo se construyen en R.
Regresión de la variable disco en peso. En la siguiente figura, usamos la función lm (modelo lineal) para encontrar la línea de regresión. Su pendiente e intersección están dadas por los valores en la ranura de coeficientes del objeto resultante reg1.
reg1 = lm(disc ~ weight, data = athletes)
a1 = reg1$coefficients[1] # intercept
b1 = reg1$coefficients[2] # slope
pline1 = ath_gg + geom_abline(intercept = a1, slope = b1,
col = "blue", lwd = 1.5)
pline1 + geom_segment(aes(xend = weight, yend = reg1$fitted),
colour = "red", arrow = arrow(length = unit(0.15, "cm")))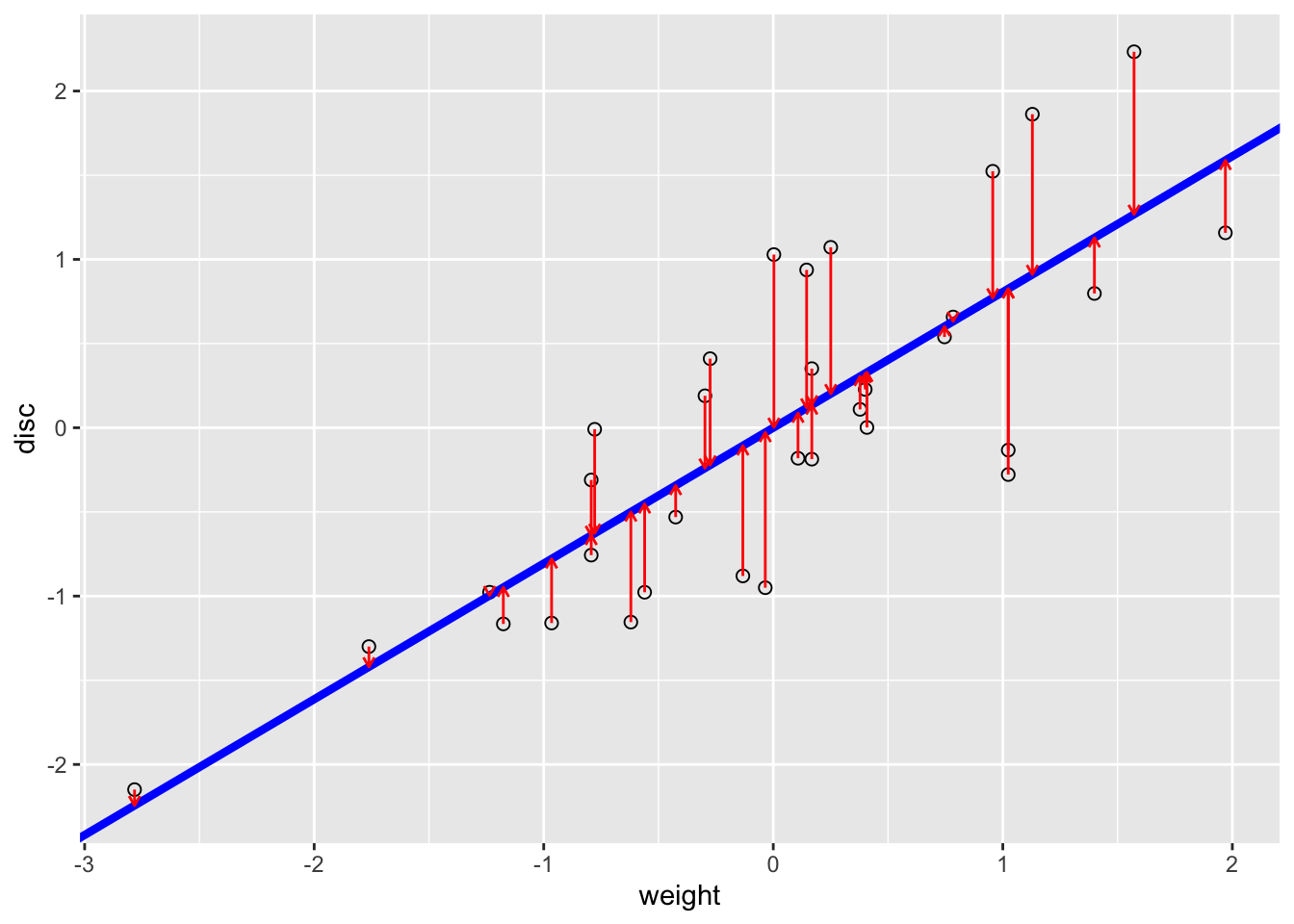 La línea azul minimiza la suma de cuadrados de los residuos verticales (en rojo).
La línea azul minimiza la suma de cuadrados de los residuos verticales (en rojo).
Regresión de peso en disco.
La siguiente figura muestra la línea producida al invertir los roles de las dos variables; el peso se convierte en la variable de respuesta.
reg2 = lm(weight ~ disc, data = athletes)
a2 = reg2$coefficients[1] # intercept
b2 = reg2$coefficients[2] # slope
pline2 = ath_gg + geom_abline(intercept = -a2/b2, slope = 1/b2,
col = "darkgreen", lwd = 1.5)
pline2 + geom_segment(aes(xend=reg2$fitted, yend=disc),
colour = "orange", arrow = arrow(length = unit(0.15, "cm")))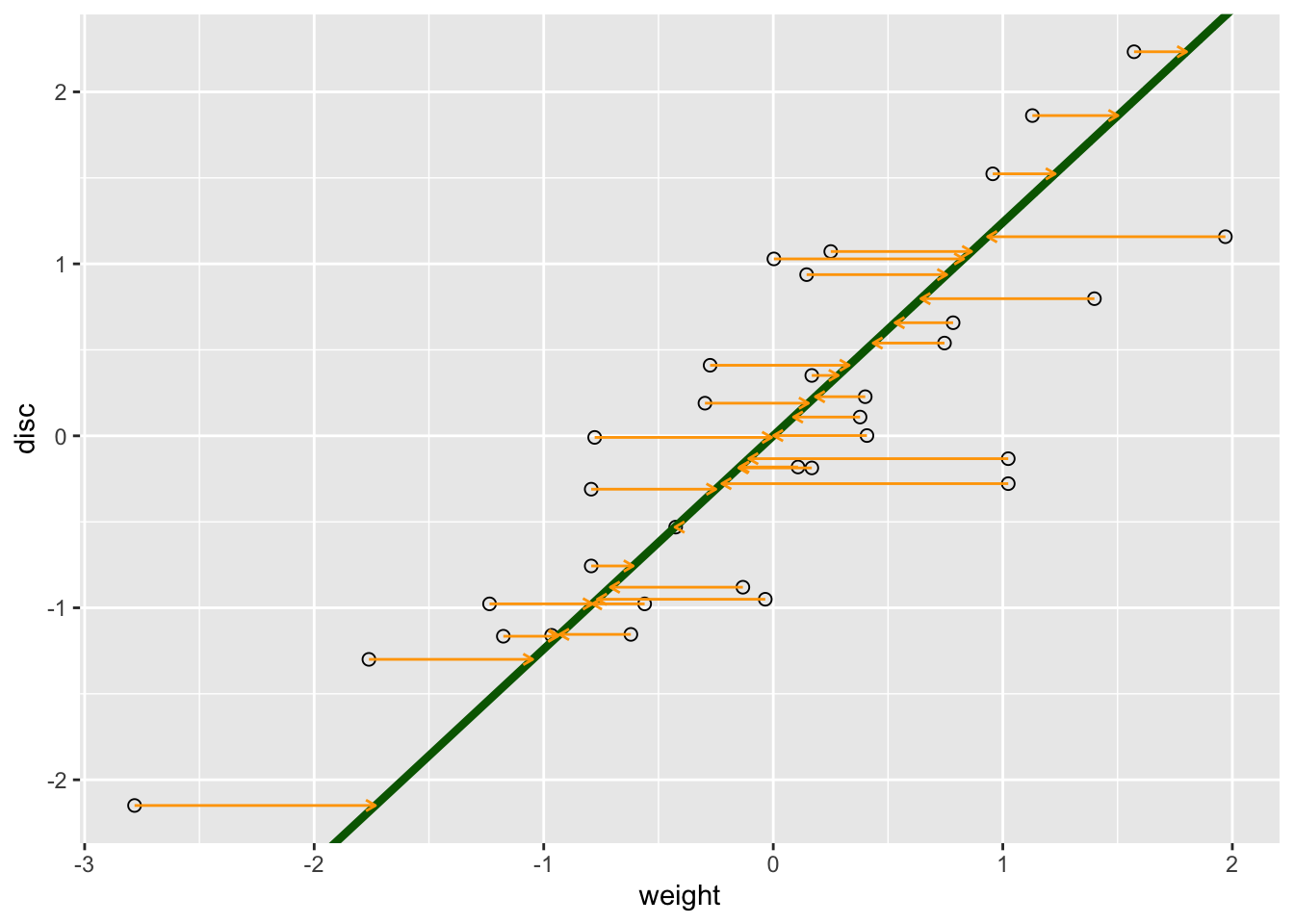 La línea verde minimiza la suma de cuadrados de los residuos horizontales (en naranja).
La línea verde minimiza la suma de cuadrados de los residuos horizontales (en naranja).
Cada una de las líneas de regresión en las figuras nos da una relación lineal aproximada entre el disco y el peso. Sin embargo, la relación difiere según cuál de las variables elijamos como predictor y cuál sea la respuesta.
8.5 El flujo de trabajo de PCA
El PCA se basa en el principio de encontrar el eje que muestra la mayor inercia / variabilidad, eliminando la variabilidad en esa dirección y luego iterando para encontrar el siguiente mejor eje ortogonal, etc. De hecho, no tenemos que ejecutar iteraciones, todos los ejes se pueden encontrar en una operación llamada Descomposición de valores singulares (profundizaremos más en los detalles a continuación).

En el diagrama de la figura, vemos que primero se calculan las medias son las varianzas y se debe elegir si trabajar con covarianzas reescaladas –la matriz de correlación– o no. Luego, el siguiente paso es la elección de k, el número de componentes relevantes para los datos. Decimos que k es el rango de la aproximación que elegimos; damos una explicación detallada de cómo tomamos esa decisión a continuación. A diferencia de la agrupación en clústeres, es imposible elegir el número de componentes antes de hacer parte del análisis. La elección de k requiere mirar un gráfico de las varianzas explicadas por los componentes principales sucesivos antes de proceder a las proyecciones de los datos.
Los resultados finales del flujo de trabajo de PCA son mapas útiles tanto de las variables como de las muestras. Comprender cómo se construyen estos mapas maximizará la información que podemos recopilar de ellos.
8.6 Graficar las observaciones en el plano principal
Ahora queremos hacer un análisis PCA completo sobre los datos de las tortugas. Recordemos que ya miramos las estadísticas de resumen para los datos de 1 y 2 dimensiones. Ahora vamos a responder a la pregunta sobre la dimensionalidad “verdadera” de estos datos reescalados. En el siguiente código, usamos la función princomp. Su valor de retorno es una lista de toda la información importante necesaria para graficar e interpretar un PCA.
cor(scaledTurtles)## length width height
## length 1.0000000 0.9783116 0.9646946
## width 0.9783116 1.0000000 0.9605705
## height 0.9646946 0.9605705 1.0000000pcaturtles = princomp(scaledTurtles)
pcaturtles## Call:
## princomp(x = scaledTurtles)
##
## Standard deviations:
## Comp.1 Comp.2 Comp.3
## 1.6954576 0.2048201 0.1448180
##
## 3 variables and 48 observations.library("factoextra")## Welcome! Want to learn more? See two factoextra-related books at https://goo.gl/ve3WBafviz_eig(pcaturtles, geom = "bar", bar_width = 0.4) + ggtitle("")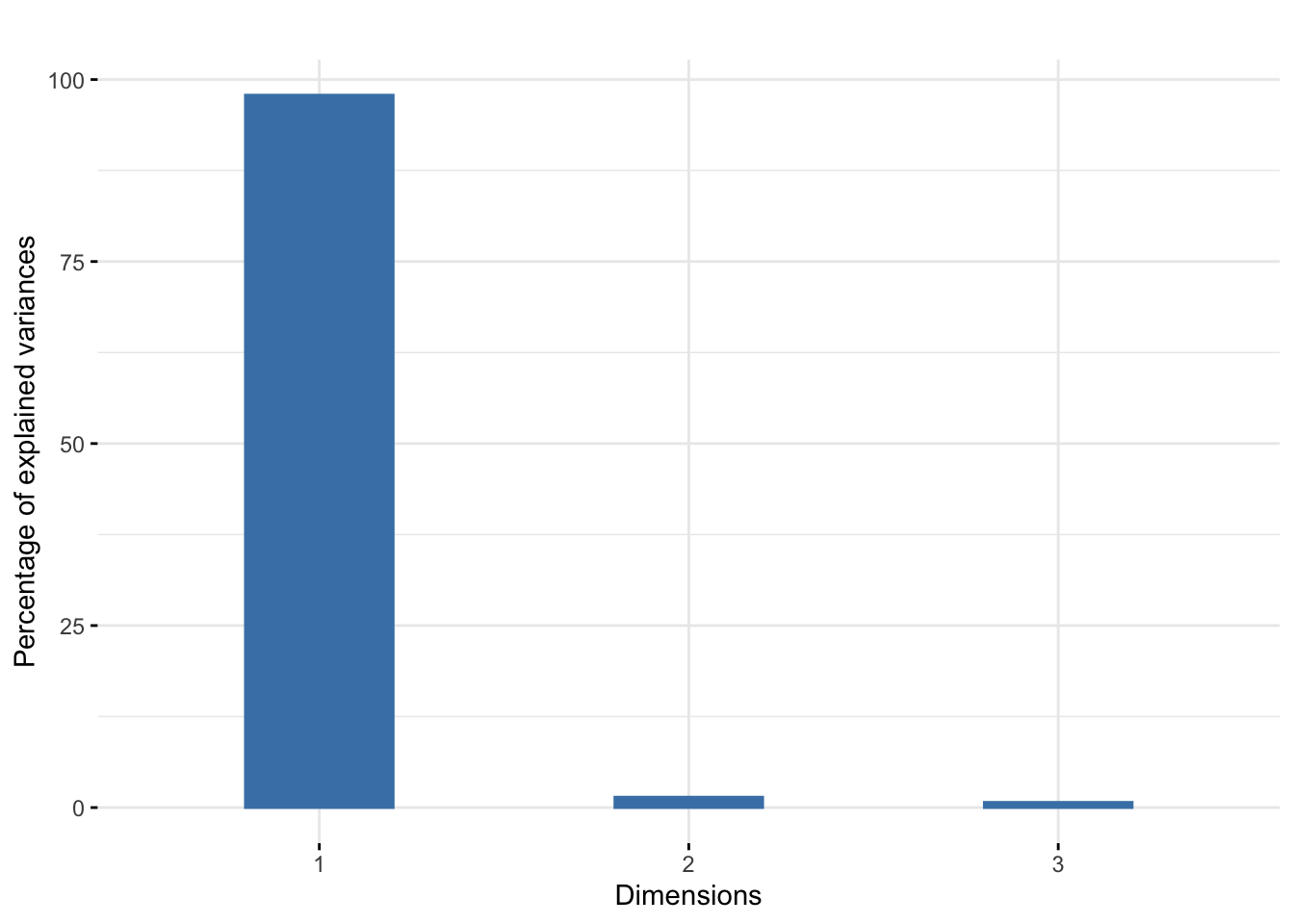
El screenplot muestra los valores propios de los datos de tortugas estandarizados (tortugas escaladas): hay un valor grande y dos pequeños. Los datos son (casi) unidimensionales. Veremos por qué esta dimensión se denomina eje de tamaño, fenómeno frecuente en los datos biométricos (Jolicoeur, Mosimann y otros 1960).
Un análisis completo: los deportistas de decatlón.
Analizamos brevemente parte de estos datos anteriormente en el práctico; aquí seguiremos paso a paso un análisis multivariado completo. Primero, echamos un segundo vistazo a la matriz de correlación redondeada que captura lo esencial de las asociaciones bivariadas.
cor(athletes) %>% round(1)## m100 long weight highj m400 m110 disc pole javel m1500
## m100 1.0 -0.5 -0.2 -0.1 0.6 0.6 0.0 -0.4 -0.1 0.3
## long -0.5 1.0 0.1 0.3 -0.5 -0.5 0.0 0.3 0.2 -0.4
## weight -0.2 0.1 1.0 0.1 0.1 -0.3 0.8 0.5 0.6 0.3
## highj -0.1 0.3 0.1 1.0 -0.1 -0.3 0.1 0.2 0.1 -0.1
## m400 0.6 -0.5 0.1 -0.1 1.0 0.5 0.1 -0.3 0.1 0.6
## m110 0.6 -0.5 -0.3 -0.3 0.5 1.0 -0.1 -0.5 -0.1 0.1
## disc 0.0 0.0 0.8 0.1 0.1 -0.1 1.0 0.3 0.4 0.4
## pole -0.4 0.3 0.5 0.2 -0.3 -0.5 0.3 1.0 0.3 0.0
## javel -0.1 0.2 0.6 0.1 0.1 -0.1 0.4 0.3 1.0 0.1
## m1500 0.3 -0.4 0.3 -0.1 0.6 0.1 0.4 0.0 0.1 1.0Luego comenzamos a mirar el screenplot, que nos ayudará a elegir un rango k para estos datos.
library(ade4)##
## Attaching package: 'ade4'## The following object is masked from 'package:Biostrings':
##
## score## The following object is masked from 'package:GenomicRanges':
##
## score## The following object is masked from 'package:BiocGenerics':
##
## scorepca.ath = dudi.pca(athletes, scannf = FALSE)
pca.ath$eig## [1] 3.4182381 2.6063931 0.9432964 0.8780212 0.5566267 0.4912275 0.4305952
## [8] 0.3067981 0.2669494 0.1018542fviz_eig(pca.ath, geom = "bar", bar_width = 0.3) + ggtitle("")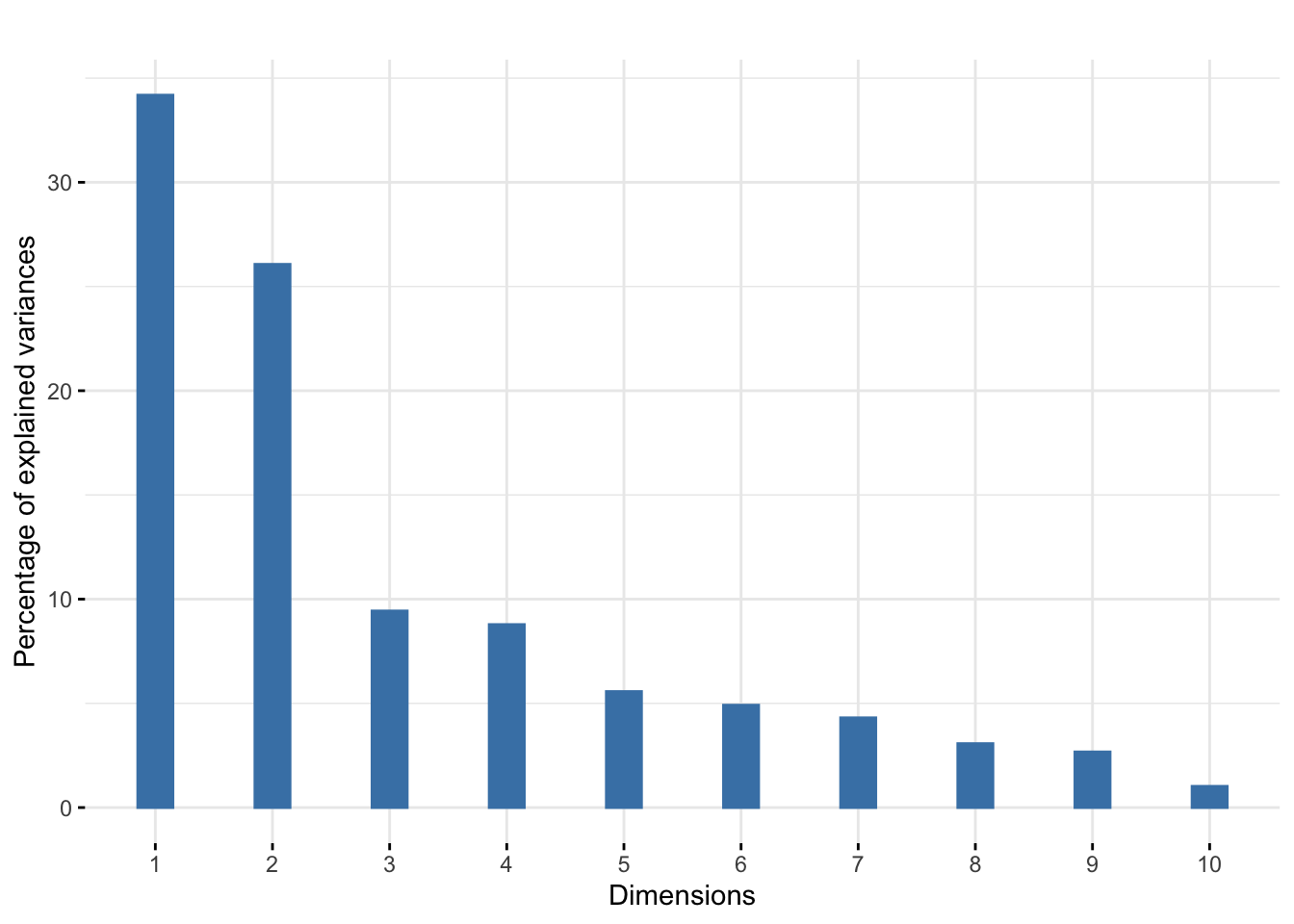
El screenplot es lo primero que debemos consultar, nos dice que es satisfactorio utilizar una gráfica bidimensional.
La figura muestra que los valores propios en el screenplot muestran una clara caída después del segundo valor propio. Esto indica que se obtendrá una buena aproximación en el rango 2. Veamos una interpretación de los dos primeros ejes proyectando las cargas de las variables originales (antiguas) a medida que se proyectan sobre las dos nuevas.
fviz_pca_var(pca.ath, col.circle = "black") + ggtitle("")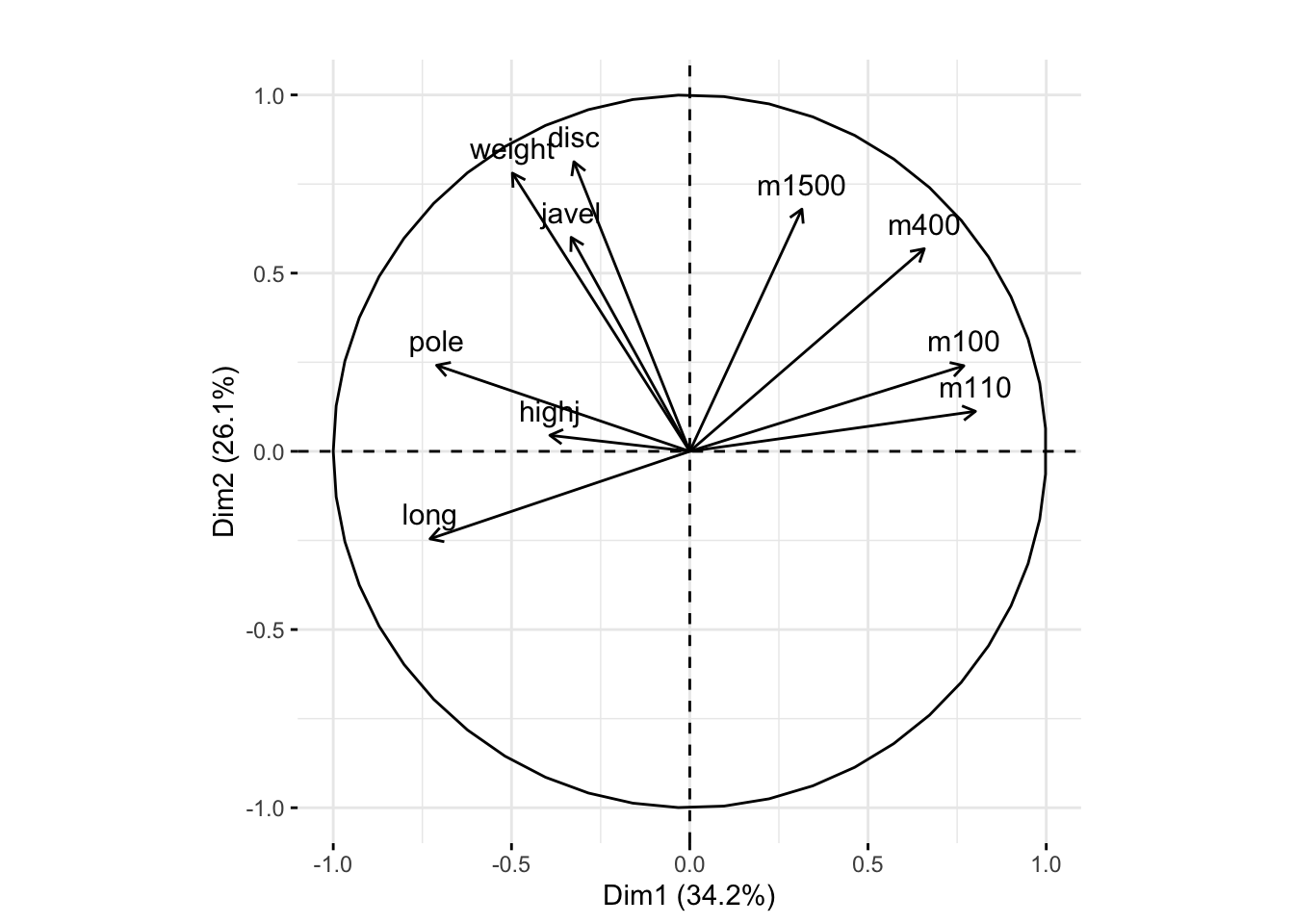 Círculo de correlación de las variables originales.
Círculo de correlación de las variables originales.
La figura muestra el círculo de correlación de la variable transformada. Ahora vemos que tenemos un eje de tamaño, todas las flechas apuntan en la misma dirección.
Ahora todas las correlaciones negativas son bastante pequeñas. El screenplot no mostrará ningún cambio, ya que los valores propios no se modifican. Los signos en los coeficientes de las cargas de PC para las m variables son la única salida que cambia.
Ahora graficamos los atletas proyectados en el primer plano principal usando:
fviz_pca_ind(pca.ath) + ggtitle("") + ylim(c(-2.5,5.7))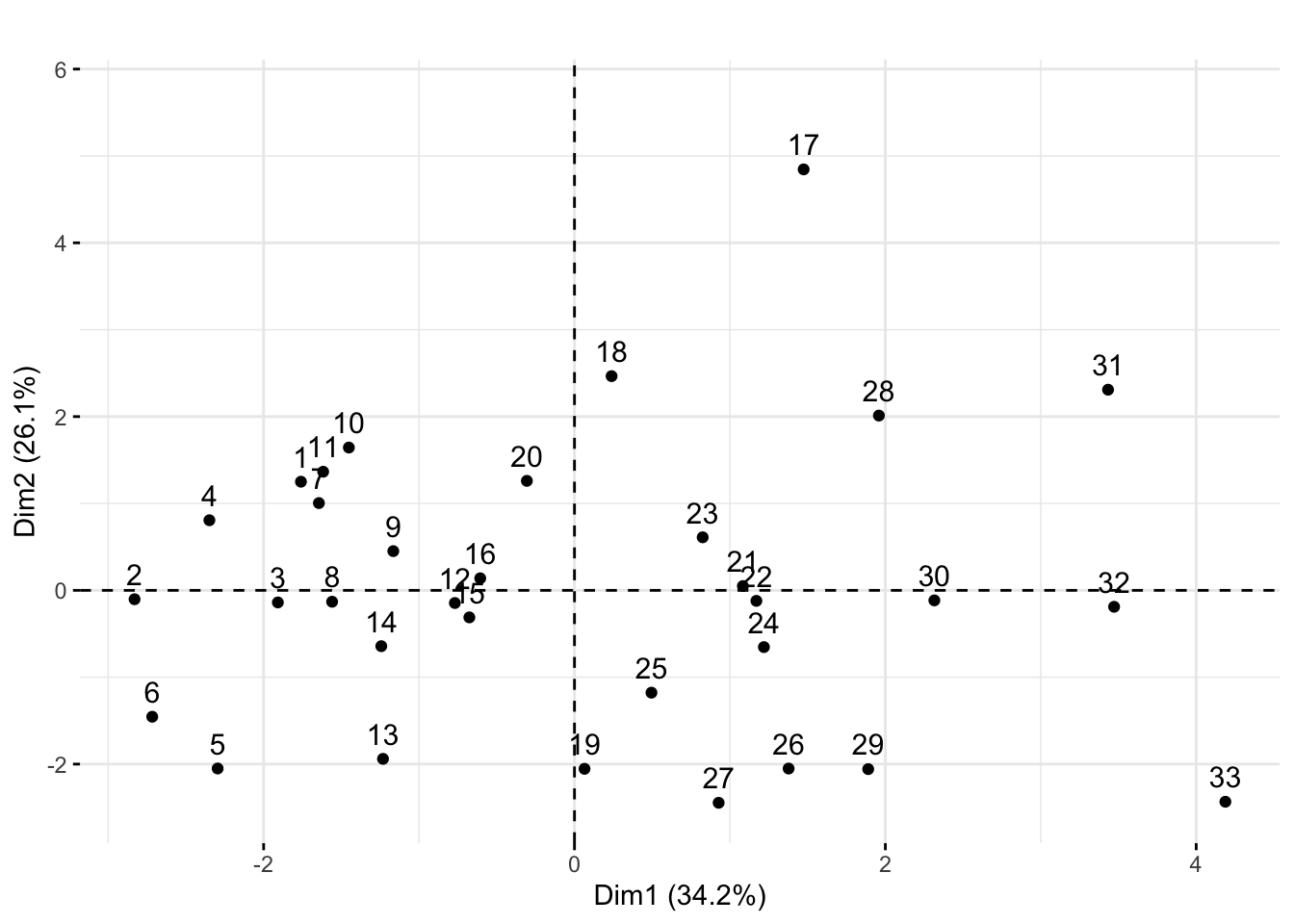
Primer plano principal que muestra las proyecciones de los atletas. ¿Notas algo sobre la organización de los números?
8.7 PCA como herramienta exploratoria: uso de información adicional
Hemos visto que, a diferencia de la regresión, PCA trata todas las variables por igual (en la medida en que fueron preprocesadas para tener desviaciones estándar equivalentes). Sin embargo, todavía es posible mapear otras variables continuas o factores categóricos en las gráficas para ayudar a interpretar los resultados. A menudo tenemos información complementaria sobre las muestras, por ejemplo, etiquetas de diagnóstico en los datos de diabetes o los tipos de células en los datos de expresión génica de células T.
Aquí vemos cómo podemos usar tales variables adicionales para informar nuestra interpretación. El mejor lugar para almacenar los llamados metadatos es en las ranuras apropiadas del objeto de datos (como en la clase Bioconductor SummarizedExperiment); el segundo mejor, en columnas adicionales de un dataframe que también contiene los datos numéricos. En la práctica, esta información se almacena a menudo de una manera más o menos críptica en los nombres de las filas de la matriz. A continuación, debemos enfrentar el último escenario, y usamos substr para extraer los tipos de células y mostrar el screenplot y el PCA en las siguientes figuras.
pcaMsig3 = dudi.pca(Msig3transp, center = TRUE, scale = TRUE,
scannf = FALSE, nf = 4)
fviz_screeplot(pcaMsig3) + ggtitle("")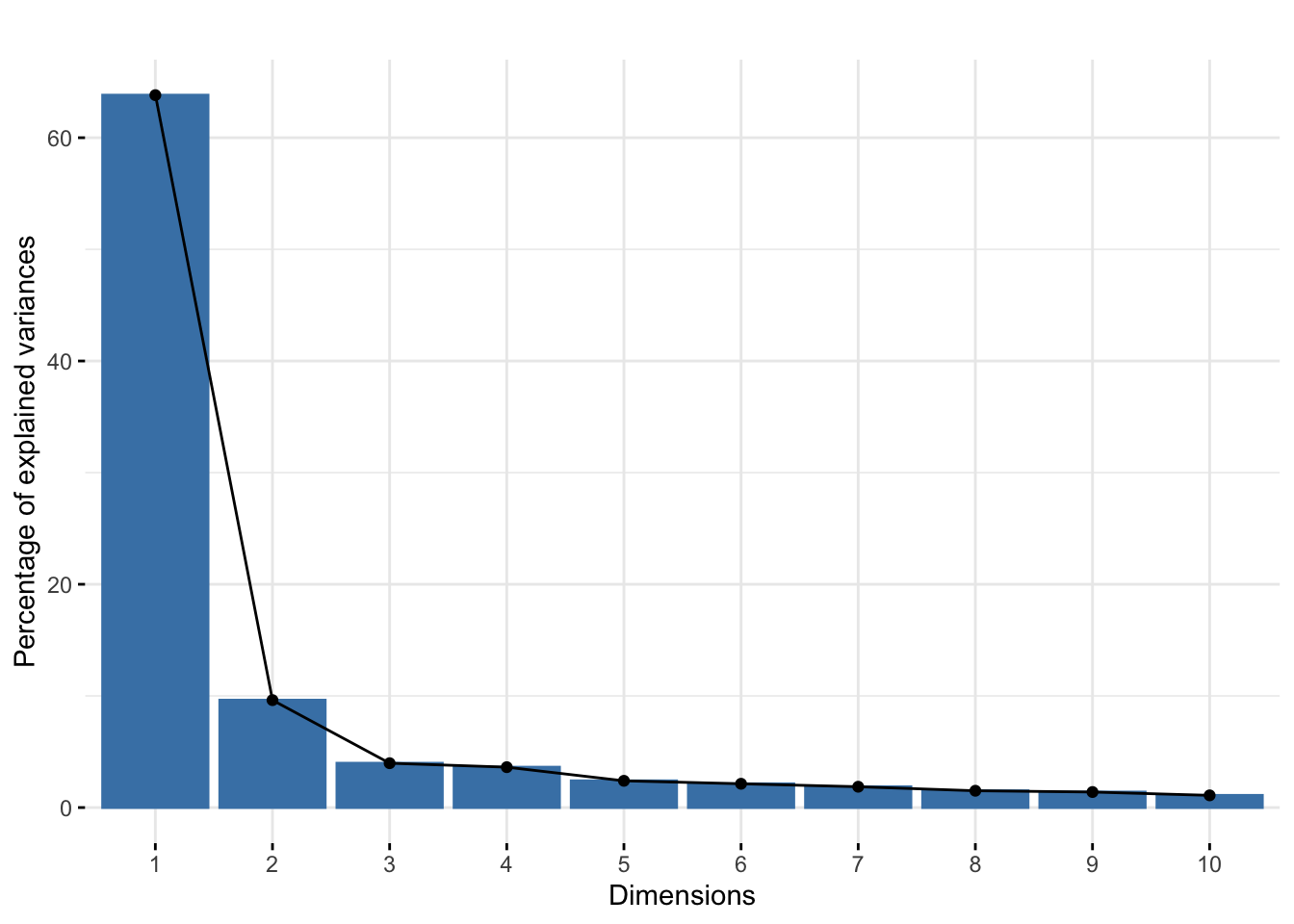
ids = rownames(Msig3transp)
celltypes = factor(substr(ids, 7, 9))
status = factor(substr(ids, 1, 3))
table(celltypes)## celltypes
## EFF MEM NAI
## 10 9 11cbind(pcaMsig3$li, tibble(Cluster = celltypes, sample = ids)) %>%
ggplot(aes(x = Axis1, y = Axis2)) +
geom_point(aes(color = Cluster), size = 5) +
geom_hline(yintercept = 0, linetype = 2) +
geom_vline(xintercept = 0, linetype = 2) +
scale_color_discrete(name = "Cluster") + coord_fixed()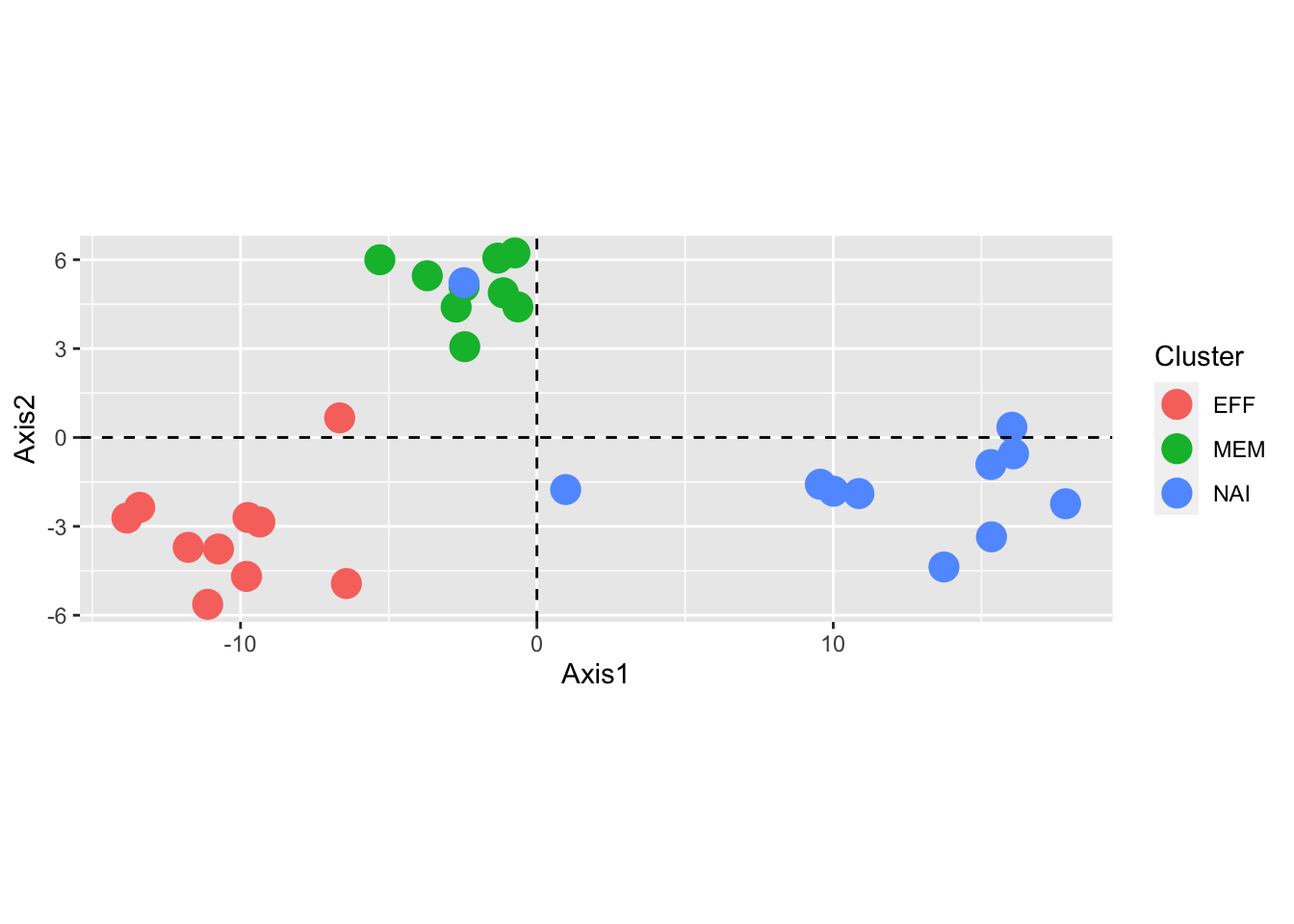
Biplots and scaling
En el ejemplo anterior, el número de variables medidas era demasiado grande para permitir un gráfico concurrente útil de variables y muestras. En este ejemplo graficamos el biplot PCA de un conjunto de datos simple donde se realizaron mediciones químicas en diferentes vinos para los cuales también tenemos una variable categórica wine.class. Comenzamos el análisis observando las correlaciones bidimensionales y un mapa de calor de las variables.
library("pheatmap")
load("data_cap8910/wine.RData")
load("data_cap8910/wineClass.RData")
wine[1:2, 1:7]## Alcohol MalicAcid Ash AlcAsh Mg Phenols Flav
## 1 14.23 1.71 2.43 15.6 127 2.80 3.06
## 2 13.20 1.78 2.14 11.2 100 2.65 2.76pheatmap(1 - cor(wine), treeheight_row = 0.2)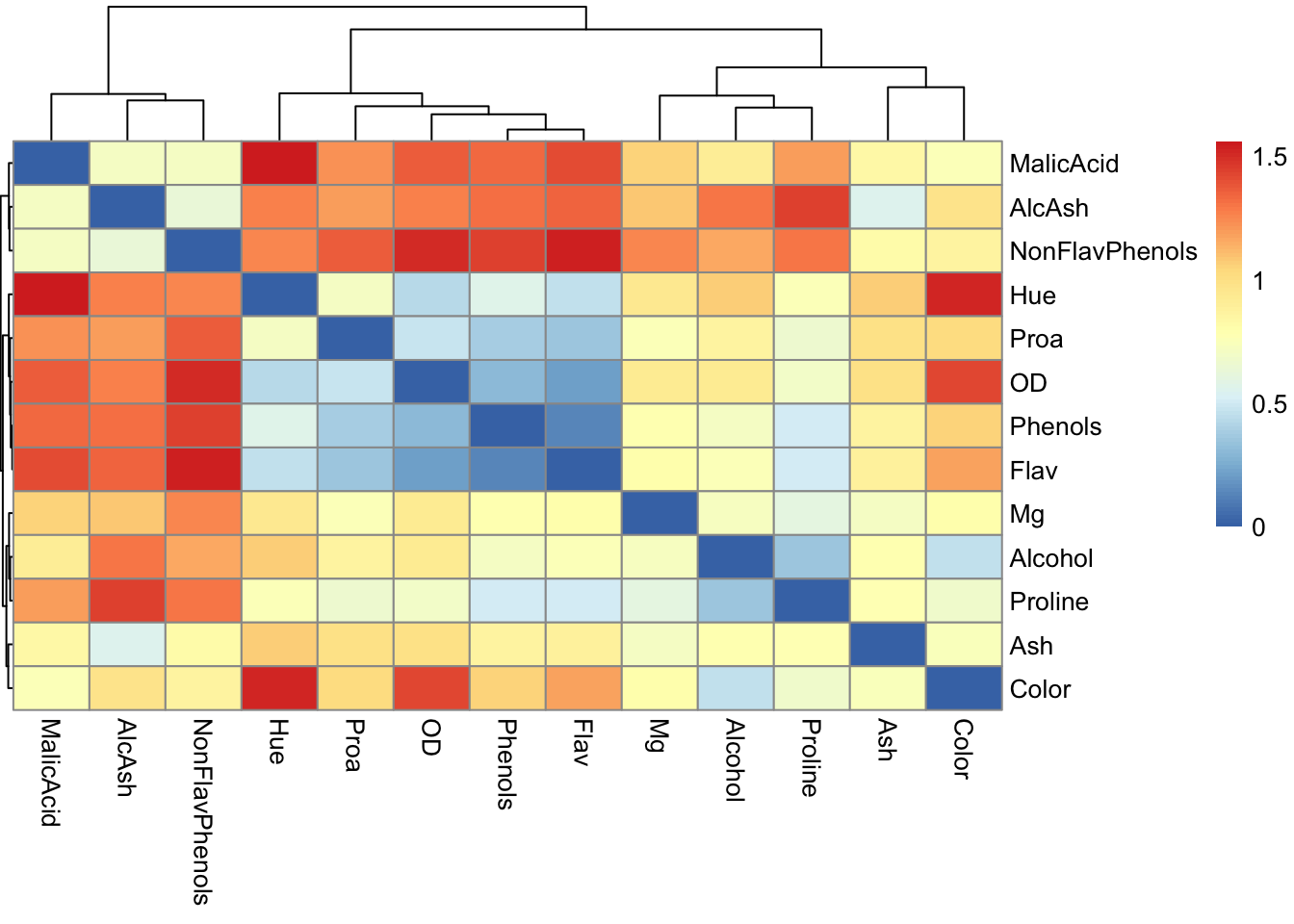
La diferencia entre 1 y la correlación se puede usar como una distancia entre variables y se usa para hacer un mapa de calor de las asociaciones entre las variables.
winePCAd = dudi.pca(wine, scannf=FALSE)
table(wine.class)## wine.class
## barolo grignolino barbera
## 59 71 48fviz_pca_biplot(winePCAd, geom = "point", habillage = wine.class,
col.var = "violet", addEllipses = TRUE, ellipse.level = 0.69) +
ggtitle("") + coord_fixed()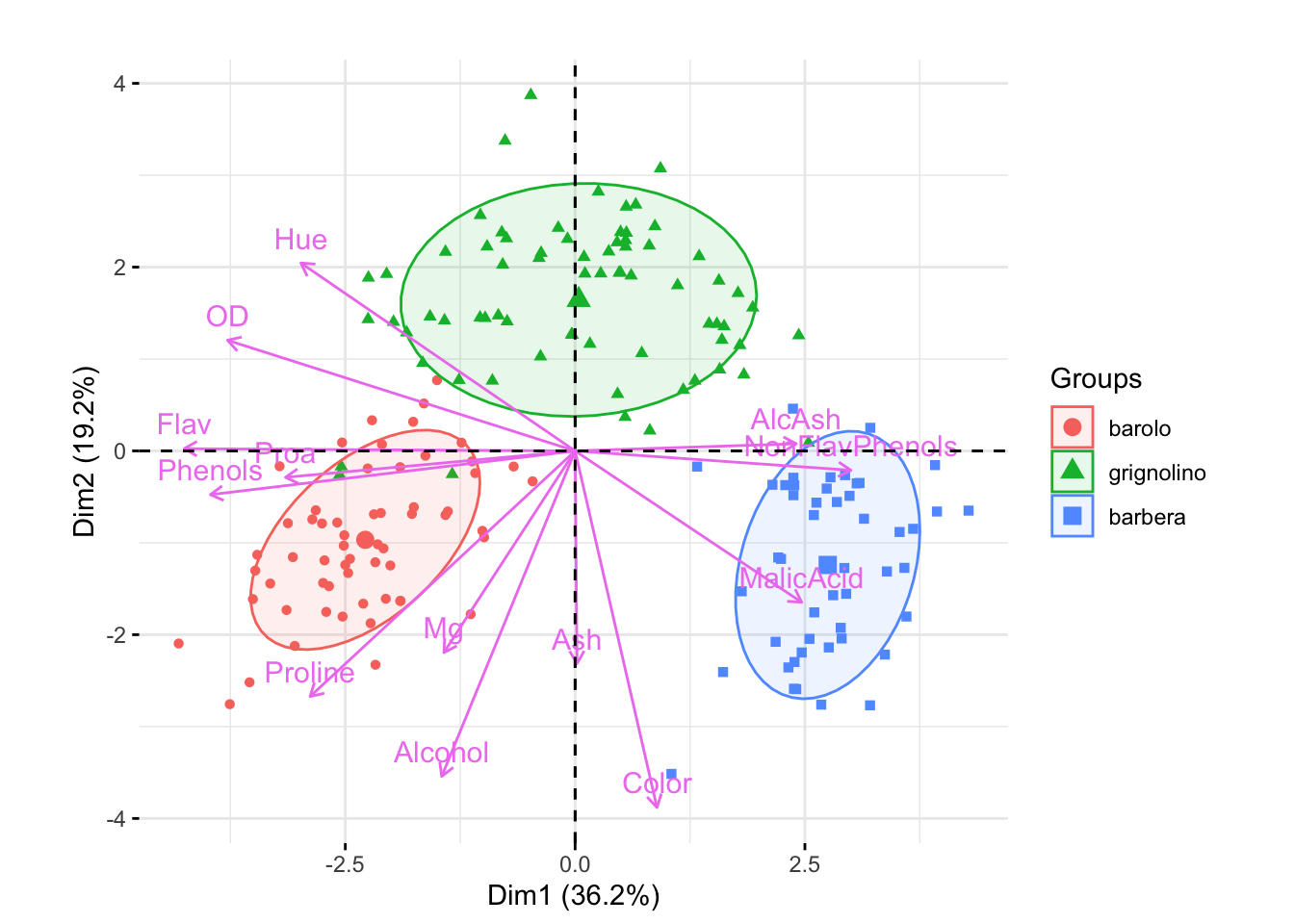
Biplot PCA que incluye elipses para los tres tipos de vino: barolo, grignolino y barbera. Para cada elipsis, las longitudes de los ejes vienen dadas por una desviación estándar. Los ángulos pequeños entre los vectores Fenoles, Flav y Proa indican que están fuertemente correlacionados, mientras que Hue y Alcohol no están correlacionados.
Una biplot es una representación simultánea tanto del espacio de observaciones como del espacio de variables. En el caso de un biplot PCA como el de la figura, las flechas representan las direcciones de las variables antiguas a medida que se proyectan sobre el plano definido por los dos primeros ejes nuevos. Aquí las observaciones son solo puntos de colores, el color se ha elegido de acuerdo con el tipo de vino que se está trazando. Podemos interpretar las direcciones de las variables con respecto a los puntos de muestra, por ejemplo, los puntos azules son del grupo barbera y muestran un mayor contenido de ácido málico que los otros vinos.
La interpretación de gráficos multivariados requiere el uso de tanta información disponible como sea posible; aquí hemos utilizado las muestras y sus grupos así como las variables para entender las principales diferencias entre los vinos.