10 Métodos multivariados para datos heterogéneos
En el práctico 8, vimos cómo resumir matrices rectangulares cuyas columnas eran variables continuas. Los mapas que hicimos utilizaron técnicas de reducción de dimensionalidad no supervisadas, como el análisis de componentes principales (PCA), destinado a aislar el componente de señal más importante en una matriz X cuando todas las columnas tienen variaciones significativas.
Aquí ampliamos estas ideas a datos heterogéneos más complejos donde se combinan datos continuos y categóricos o incluso a datos donde las variables individuales no están disponibles. De hecho, a veces nuestras observaciones no pueden describirse fácilmente por características, pero es posible determinar distancias o (des) similitudes entre ellas, o ponerlas en un gráfico o un árbol. Los ejemplos incluyen especies en un árbol de especies o secuencias biológicas. Fuera de la biología, existen documentos de texto o archivos de sonido, donde podemos tener un método razonable para determinar la (des) similitud entre objetos, pero ningún “sistema de coordenadas” absoluto de características.
Este capítulo contiene técnicas más avanzadas para las que hemos omitido muchos detalles técnicos. Esperamos que obtengan alguna experiencia práctica con ejemplos y referencias extensas. Esperamos que comprendan algunas de las técnicas más “de vanguardia” en los análisis multivariados no lineales.
10.1 Escalado y ordenación multidimensionales
A veces, los datos no se representan como puntos en un espacio de características. Esto puede ocurrir cuando se nos proporcionan matrices de (dis) similitud entre objetos como drogas, imágenes, árboles u otros objetos complejos, que no tienen coordenadas obvias en \(Rˆ{n}\).
Aquí nuestro objetivo es visualizar los datos en mapas en espacios de baja dimensión (por ejemplo, planos) que recuerdan a los que hacemos desde los primeros ejes principales en PCA.
Comenzamos con un ejemplo que muestra lo que podemos hacer con datos geográficos simples. En la figura a continuación se muestra un heatmap y la agrupación de las distancias aproximadas de las carreteras entre algunas de las ciudades europeas.
library("pheatmap")
load("data_cap8910/distEuroN.RData")
seteuro = as.matrix(distEuroN)[1:12, 1:12]
pheatmap(seteuro, cluster_rows = TRUE,
treeheight_row = 0.0001, treeheight_col = 0.8,
fontsize_col = 8, cellwidth = 13, cellheight = 13)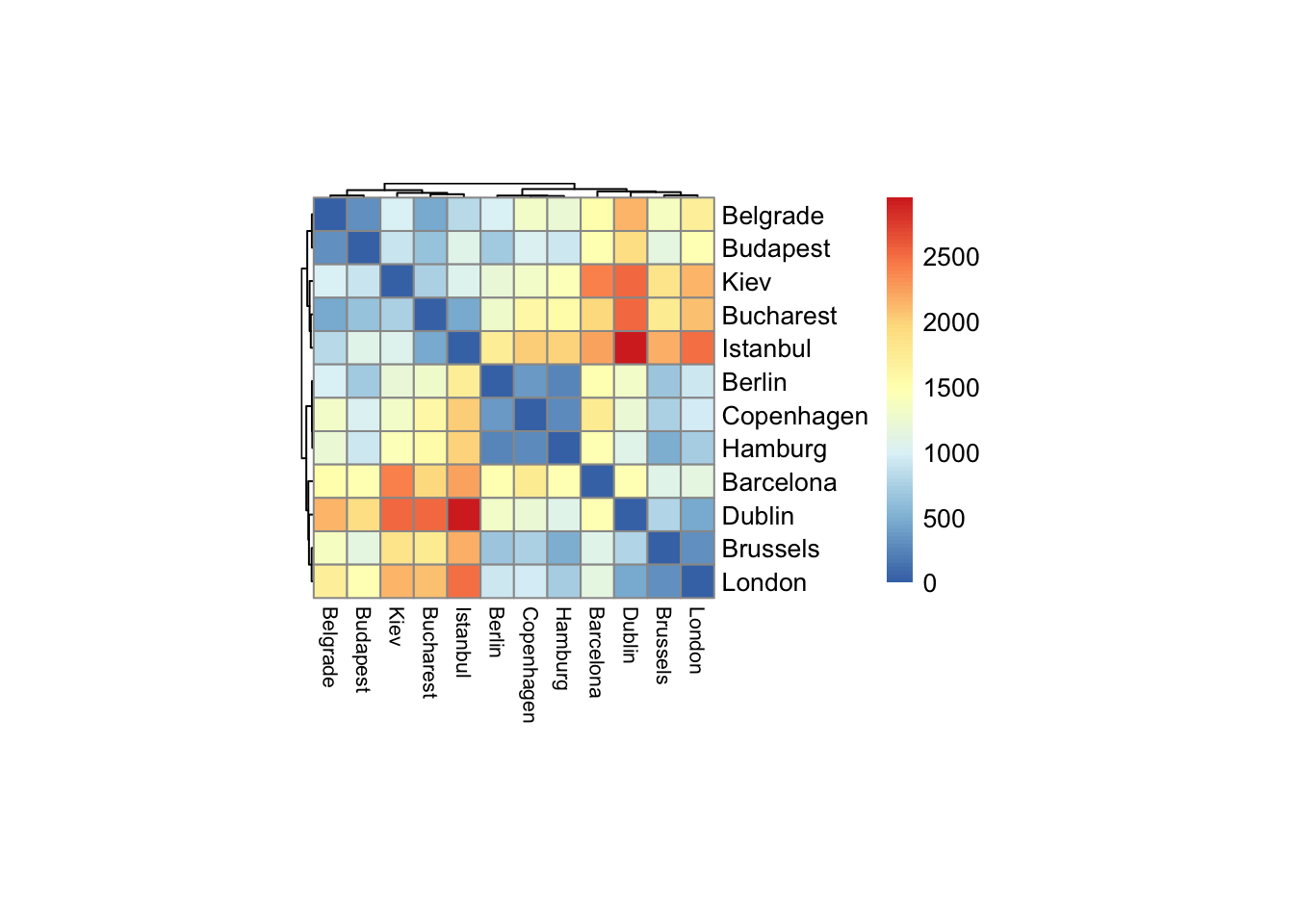
Un heatmap de las distancias entre algunas de las ciudades. La función ha reordenado el orden de las ciudades, agrupando las más cercanas.
Dadas estas distancias entre ciudades, la escala multidimensional (MDS) proporciona un “mapa” de sus ubicaciones relativas. Por supuesto, en este caso, las distancias se midieron originalmente como distancias por carretera (excepto para transbordadores), por lo que en realidad esperamos encontrar un mapa bidimensional que represente bien los datos. Con datos biológicos, es probable que nuestros mapas sean menos claros. Llamamos a la función con:
MDSEuro = cmdscale(distEuroN, eig = TRUE)Hacemos una función que podemos reutilizar para hacer el screeplot de MDS a partir del resultado de una llamada a la función cmdscale:
library("tibble")
plotbar = function(res, m = 9) {
tibble(eig = res$eig[seq_len(m)], k = seq(along = eig)) %>%
ggplot(aes(x = k, y = eig)) +
scale_x_discrete("k", limits = seq_len(m)) + theme_minimal() +
geom_bar(stat="identity", width=0.5, color="orange", fill="pink")
}
plotbar(MDSEuro, m = 5)## Warning: Continuous limits supplied to discrete scale.
## Did you mean `limits = factor(...)` or `scale_*_continuous()`?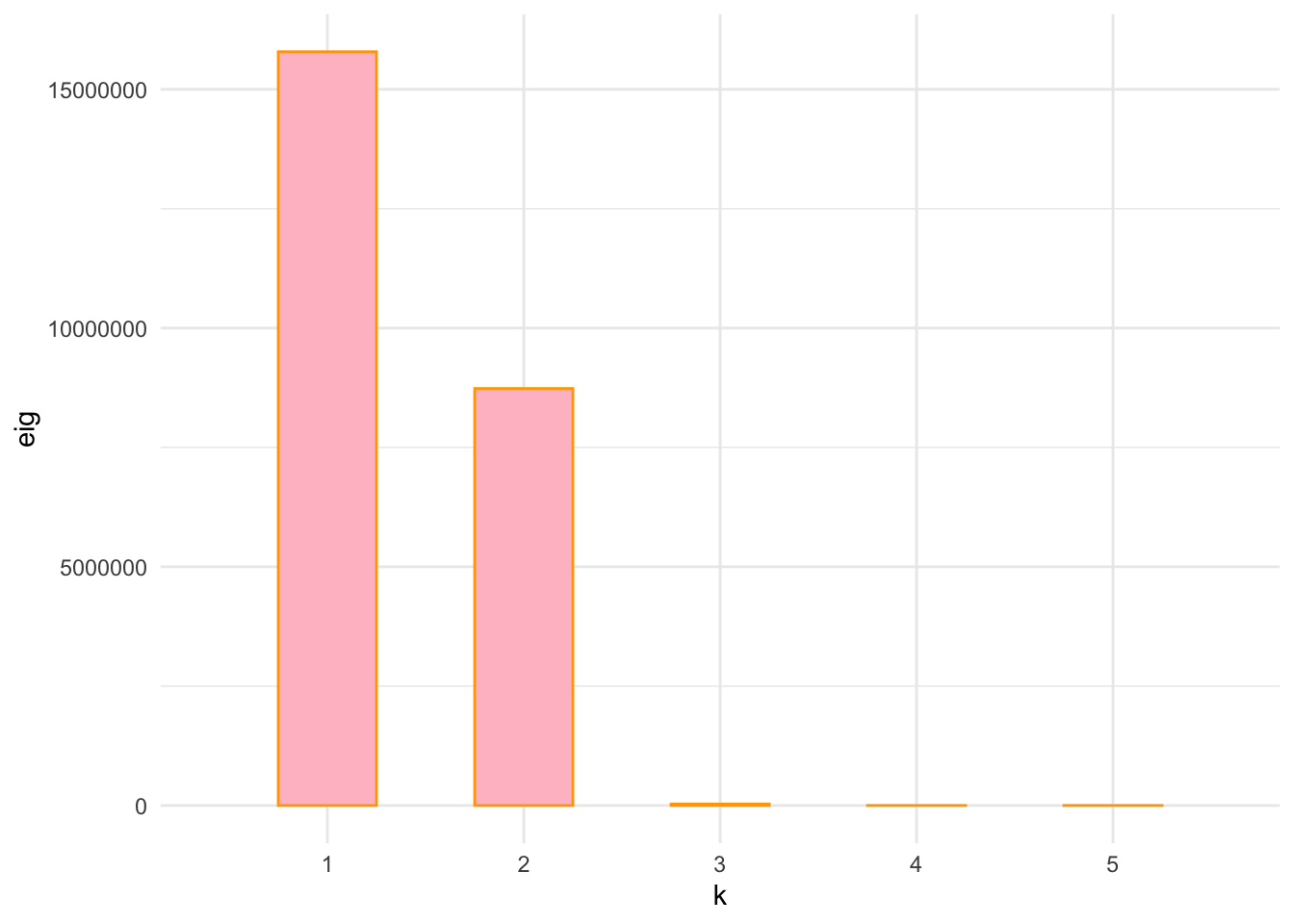
Screeplot de los primeros 5 valores propios. La caída después de los dos primeros valores propios es muy visible.
Principal Coordinates Análisis (PCoA)
PCoA, conocido también como escalado métrico multidimensional (MDS) es conceptualmente similar a PCA y análisis de correspondencia (CA) que preservan distancias Eudlicean y chi-cuadrado entre objetos, respectivamente. La diferencia con estos métodos de ordenación es que el PCoA puede preservar las distancias generadas a partir de cualquier medida de similitud o disimilitud permitiendo un manejo más flexible de datos ecológicos complejos. PCA se usa comúnmente para similitudes y PCoA para diferencias.
Una ventaja importante es que el PCoA permite manejar matrices de disimilitud calculadas a partir de variables cuantitativas, semicuantitativas, cualitativas y mixtas. En este caso la elección de la medida de similitud o disimilitud es crítica y debe ser adecuada para los datos con los que se está trabajando.
Aunque, este método presenta varias ventajas hay que recordar que el PCoA representa en el plano los componentes euclidianos de la matriz, incluso si la matriz contiene distancias no euclidianas.
Encontrar la dimensionalidad subyacente correcta.
Como ejemplo, tomemos objetos para los que tenemos similitudes (sustitutos de distancias) pero para los que no existe un espacio euclidiano subyacente natural. En un experimento de psicología de la década de 1950, Ekman (1954) pidió a 31 sujetos que clasificaran las similitudes de 14 colores diferentes. Su objetivo era comprender la dimensionalidad subyacente de la percepción del color. La matriz de similitud o confusión fue escalada para tener valores entre 0 y 1. Los colores que a menudo se confundían tenían similitudes cercanas a 1. Transformamos los datos en una disimilitud restando los valores de 1:
ekm = read.table("data_cap8910//ekman.txt", header=TRUE)
rownames(ekm) = colnames(ekm)
disekm = 1 - ekm - diag(1, ncol(ekm))
disekm[1:5, 1:5]## w434 w445 w465 w472 w490
## w434 0.00 0.14 0.58 0.58 0.82
## w445 0.14 0.00 0.50 0.56 0.78
## w465 0.58 0.50 0.00 0.19 0.53
## w472 0.58 0.56 0.19 0.00 0.46
## w490 0.82 0.78 0.53 0.46 0.00disekm = as.dist(disekm)Calculamos las coordenadas MDS y los valores propios (eigenvalues). Combinamos los valores propios en el screeplot que se muestra en la siguiente figura:
mdsekm = cmdscale(disekm, eig = TRUE)
plotbar(mdsekm)## Warning: Continuous limits supplied to discrete scale.
## Did you mean `limits = factor(...)` or `scale_*_continuous()`?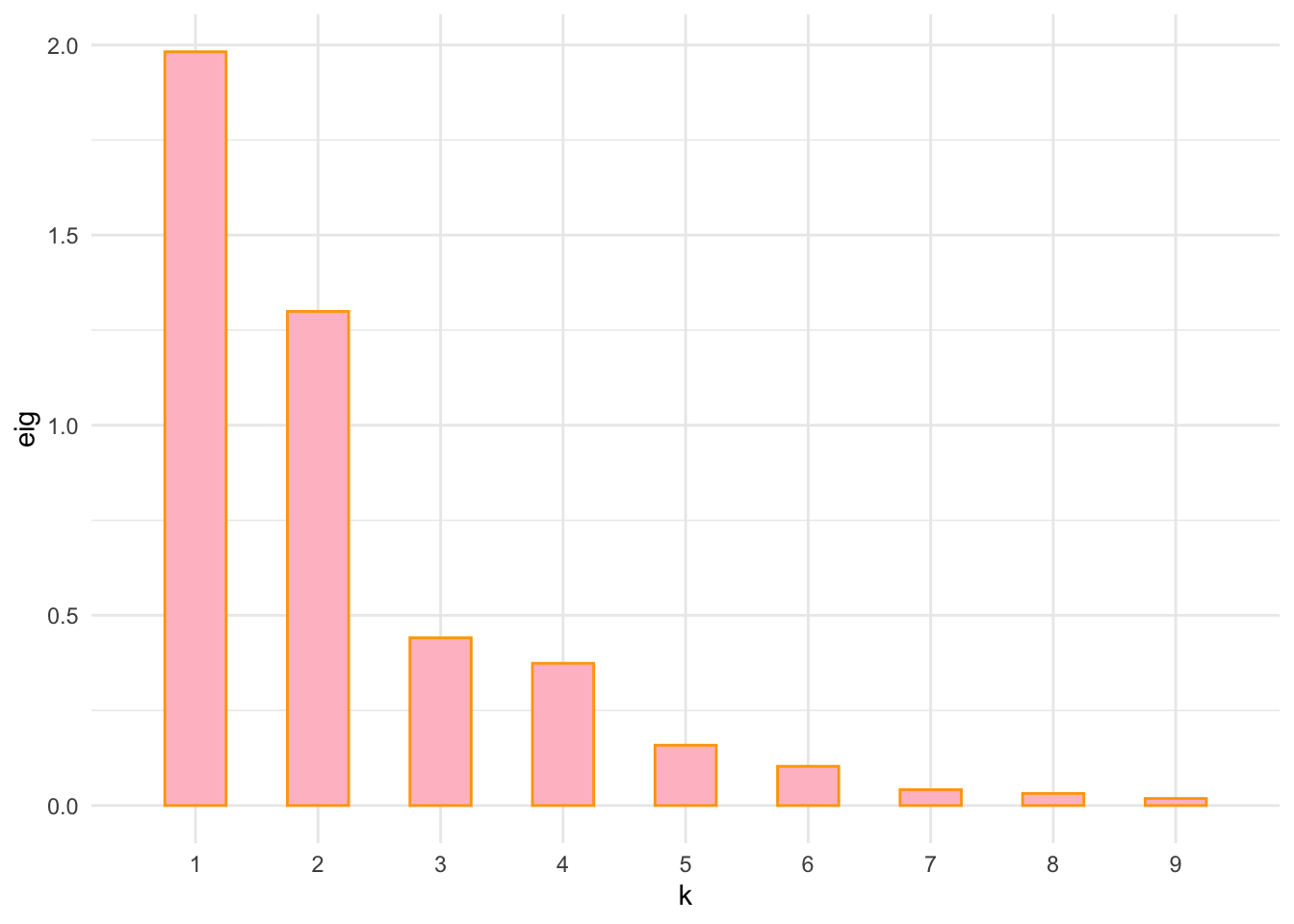
El screeplot nos muestra que el fenómeno es bidimensional, dando una respuesta clara a la pregunta de Ekman.
Graficamos los diferentes colores usando las dos primeras coordenadas principales de la siguiente manera:
dfekm = as_tibble(mdsekm$points[,1:2], .name_repair = "minimal") %>%
setNames(paste0("MDS", 1:2)) %>%
mutate(
name = rownames(ekm),
rgb = photobiology::w_length2rgb(
as.numeric(sub("w", "", name))))
library("ggrepel")
ggplot(dfekm, aes(x = MDS1, y = MDS2)) +
geom_point(col = dfekm$rgb, size = 4) +
geom_text_repel(aes(label = name)) + coord_fixed()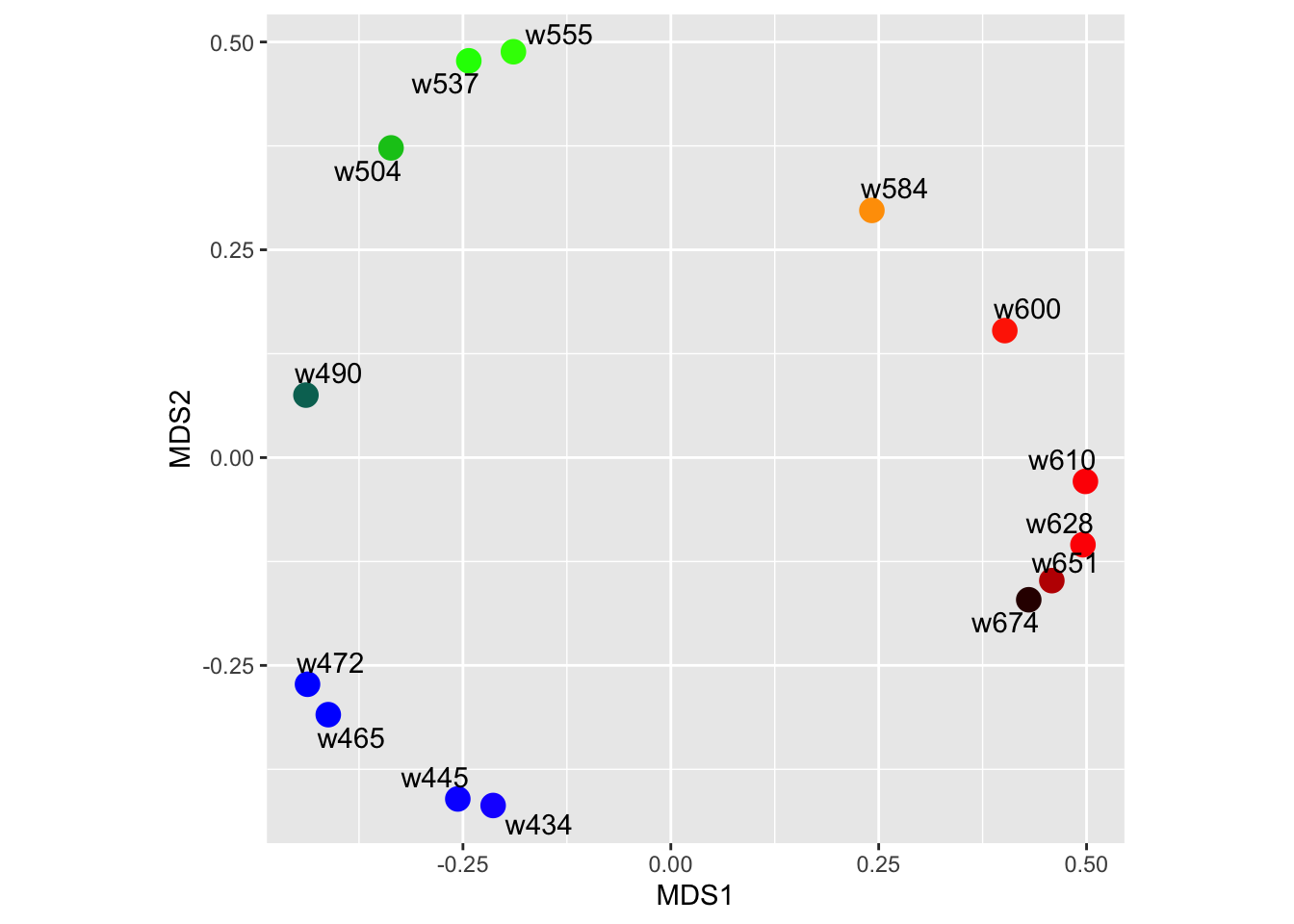
El diseño de los puntos de dispersión en las dos primeras dimensiones tiene forma de herradura. Las etiquetas y los colores muestran que el arco corresponde a las longitudes de onda.
La figura muestra los datos de Ekman en las nuevas coordenadas. Hay un patrón sorprendente que requiere explicación. Esta estructura de herradura o arco en los puntos es a menudo un indicador de un orden o gradiente latente secuencial en los datos (Diaconis, Goel y Holmes 2007).
10.2 Información contigua o complementaria
En el práctico 4 presentamos la clase R data.frame que nos permite combinar tipos de datos heterogéneos: factores categóricos, texto y valores continuos. Cada fila de un dataframe corresponde a un objeto o registro, y las columnas son las diferentes variables o características.
La información adicional sobre lotes de muestras, fechas de medición, diferentes protocolos a menudo se denominan metadatos; esto puede ser un nombre inapropiado si se da a entender que los metadatos son de alguna manera menos importantes. Dicha información son datos reales que deben integrarse en los análisis. Por lo general, lo almacenamos en un dataframe o una clase R similar y lo vinculamos estrechamente a los datos del ensayo primario.
Lotes conocidos en datos
Aquí mostramos un ejemplo de un análisis realizado por Holmes et al. (2011) sobre datos de abundancia bacteriana de microarrays Phylochip (Brodie et al. 2006). El experimento fue diseñado para detectar diferencias entre un grupo de ratas sanas y un grupo que tenía la enfermedad del intestino irritable (Nelson et al. 2010). Este ejemplo muestra un caso en el que los efectos molestos de los lotes (cuisance batch effects) se hacen evidentes en el análisis de datos experimentales. Es una ilustración del hecho de que las mejores prácticas en el análisis de datos son secuenciales y que es mejor analizar los datos a medida que se recopilan para ajustar los problemas graves en el diseño experimental a medida que ocurren, en lugar de tener que lidiar con ellos post mortem.
Cuando se inició la recolección de datos en este proyecto, se entregaron los días 1 y 2 y realizamos la trama que aparece en la figura que construiremos a continuación. Esto mostró un efecto de día definido. Al investigar la fuente de este efecto, encontramos que tanto el protocolo como la matriz eran diferentes en los días 1 y 2. Esto conduce a la incertidumbre en la fuente de variación, lo que llamamos efectos de confusión (confunding of effects).
Cargamos los datos y los paquetes que usamos para esta sección:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("sva")IBDchip = readRDS("data_cap8910//vsn28Exprd.rds")
library("ade4")
library("factoextra")
library("sva")## Loading required package: mgcv## Loading required package: nlme##
## Attaching package: 'nlme'## The following object is masked from 'package:flowWorkspace':
##
## getData## The following object is masked from 'package:Biostrings':
##
## collapse## The following object is masked from 'package:IRanges':
##
## collapse## The following object is masked from 'package:dplyr':
##
## collapse## This is mgcv 1.8-36. For overview type 'help("mgcv-package")'.##
## Attaching package: 'mgcv'## The following object is masked from 'package:gtools':
##
## scat## Loading required package: BiocParallel¿Qué clase es el IBDchip? Mira la última fila de la matriz, ¿qué notas?
class(IBDchip)## [1] "matrix" "array"dim(IBDchip)## [1] 8635 28tail(IBDchip[,1:3])## 20CF 20DF 20MF
## bm-026.1.sig_st 7.299308 7.275802 7.383103
## bm-125.1.sig_st 8.538857 8.998562 9.296096
## bru.tab.d.HIII.Con32.sig_st 6.802736 6.777566 6.859950
## bru.tab.d.HIII.Con323.sig_st 6.463604 6.501139 6.611851
## bru.tab.d.HIII.Con5.sig_st 5.739235 5.666060 5.831079
## day 2.000000 2.000000 2.000000summary(IBDchip[nrow(IBDchip),])## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 1.000 1.000 2.000 1.857 2.000 3.000Los datos son mediciones de abundancia normalizadas de 8634 taxones medidos en 28 muestras. Usamos una transformación de umbral de rango, dando a los 3000 puntajes de taxas más abundantes de 3000 a 1, y dejando que los 5634 menos abundantes tengan todos un puntaje de 1. Separamos los datos del ensayo de la variable del día, que debe considerarse un factor :
assayIBD = IBDchip[-nrow(IBDchip), ]
day = factor(IBDchip[nrow(IBDchip), ])En lugar de utilizar los datos continuos y normalizados, utilizamos un análisis robusto que reemplaza los valores por sus rangos. Los valores más bajos se consideran vínculos codificados como un umbral elegido para reflejar el número de taxones esperados que se cree que están presentes:
rankthreshPCA = function(x, threshold = 3000) {
ranksM = apply(x, 2, rank)
ranksM[ranksM < threshold] = threshold
ranksM = threshold - ranksM
dudi.pca(t(ranksM), scannf = FALSE, nf = 2)
}pcaDay12 = rankthreshPCA(assayIBD[,day!=3])
day12 = day[day!=3]
fviz(pcaDay12, element="ind", axes=c(1,2), geom=c("point","text"),
habillage = day12, repel = TRUE, palette = "Dark2",
addEllipses = TRUE, ellipse.type = "convex") + ggtitle("") +
coord_fixed()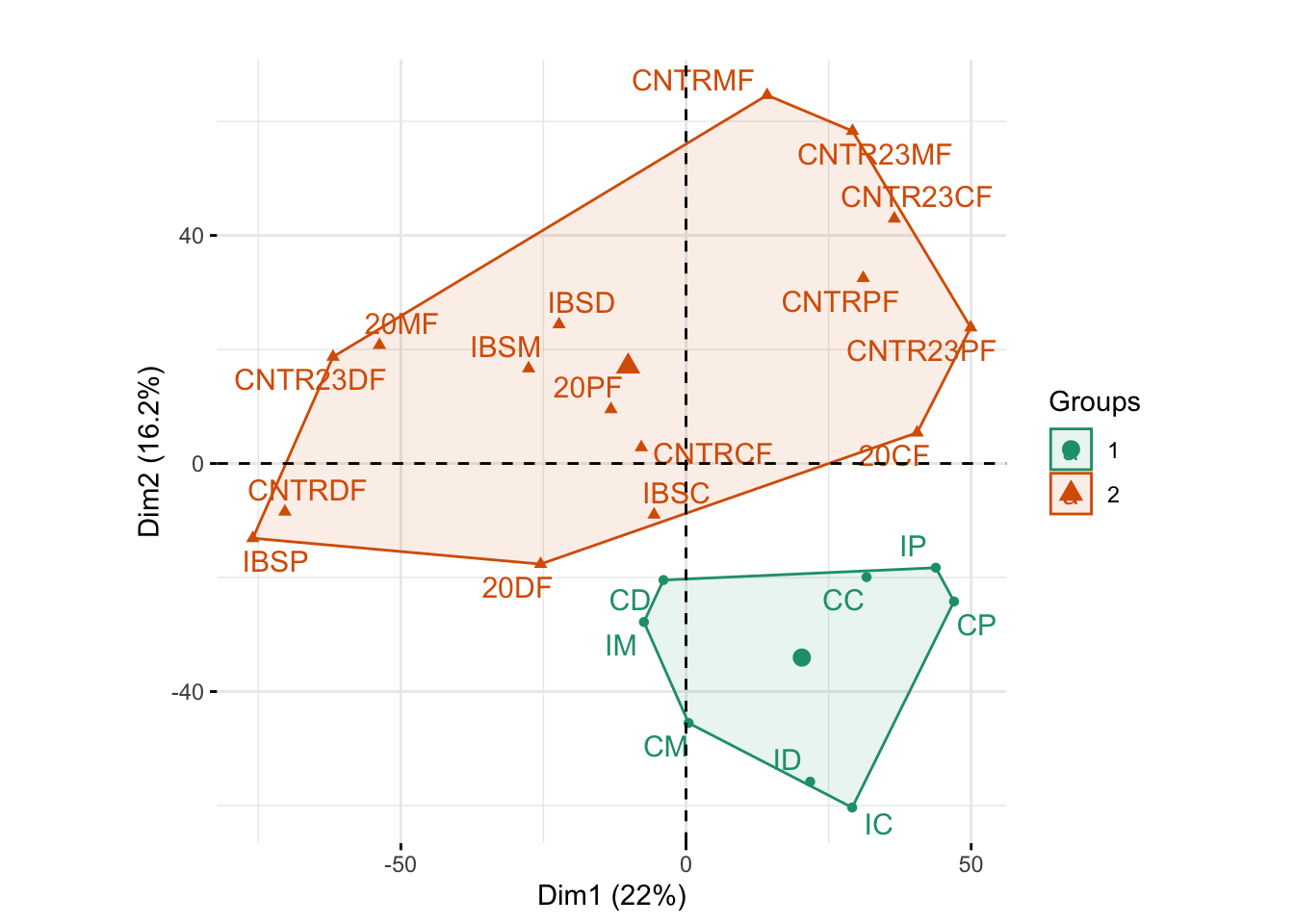
Hemos utilizado colores para identificar los diferentes días y también hemos conservado las etiquetas de muestra. También hemos agregado cascos convexos para cada día. La media del grupo se identifica como el punto con el símbolo más grande (círculo, triángulo o cuadrado).
¿Por qué usamos un umbral para los rangos?
Las abundancias bajas, a nivel de ruido, se dan para especies que realmente no están presentes, de las cuales hay más de la mitad. Un gran salto en el rango de estas observaciones podría ocurrir fácilmente sin ninguna razón significativa. De esta manera creamos una gran cantidad de vínculos de baja abundancia.
La figura anterior muestra que la muestra se organiza naturalmente en dos grupos diferentes según el día de las muestras. Después de descubrir este efecto, profundizamos en las diferencias que podrían explicar estos distintos grupos. Se utilizaron dos protocolos diferentes (protocolo 1 el día 1, protocolo 2 el día 2) y, lamentablemente, dos procedencias diferentes para las matrices utilizadas en esos dos días (matriz 1 el día 1, matriz 2 el día 2).
Se tuvo que recopilar un tercer conjunto de datos de cuatro muestras para deconvolver el efecto de confusión. La matriz 2 se utilizó con el protocolo 2 el día 3. La siguiente figura muestra la nueva gráfica de PCA con todas las muestras creadas por lo siguiente:
pcaDay123 = rankthreshPCA(assayIBD)
fviz(pcaDay123, element="ind", axes=c(1,2), geom=c("point","text"),
habillage = day, repel=TRUE, palette = "Dark2",
addEllipses = TRUE, ellipse.type = "convex") + ggtitle("") +
coord_fixed()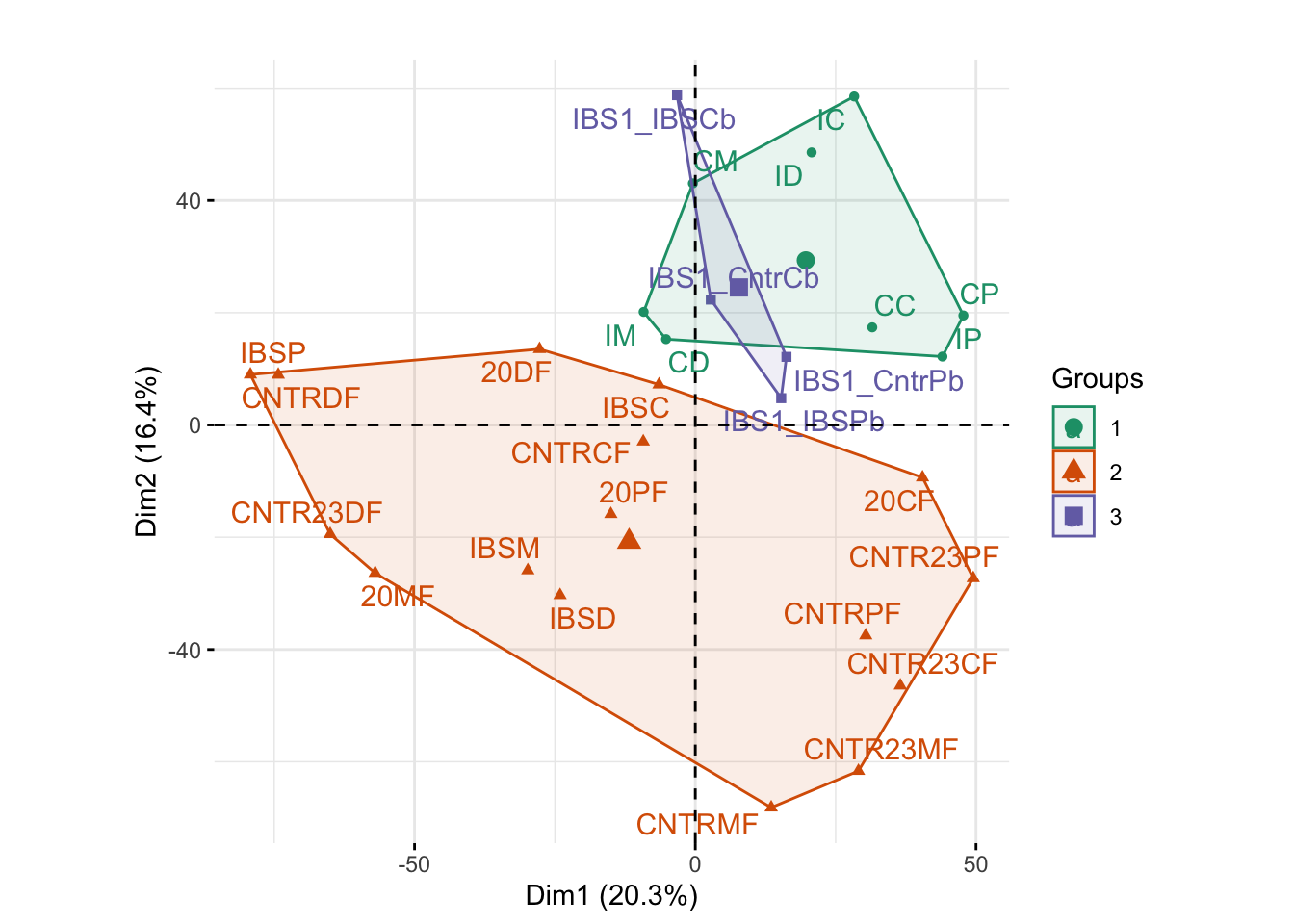
¿En qué situación sería preferible hacer elipses de confianza alrededor de los medios de grupo usando el siguiente código?
fviz_pca_ind(pcaDay123, habillage = day, labelsize = 3,
palette = "Dark2", addEllipses = TRUE, ellipse.level = 0.69)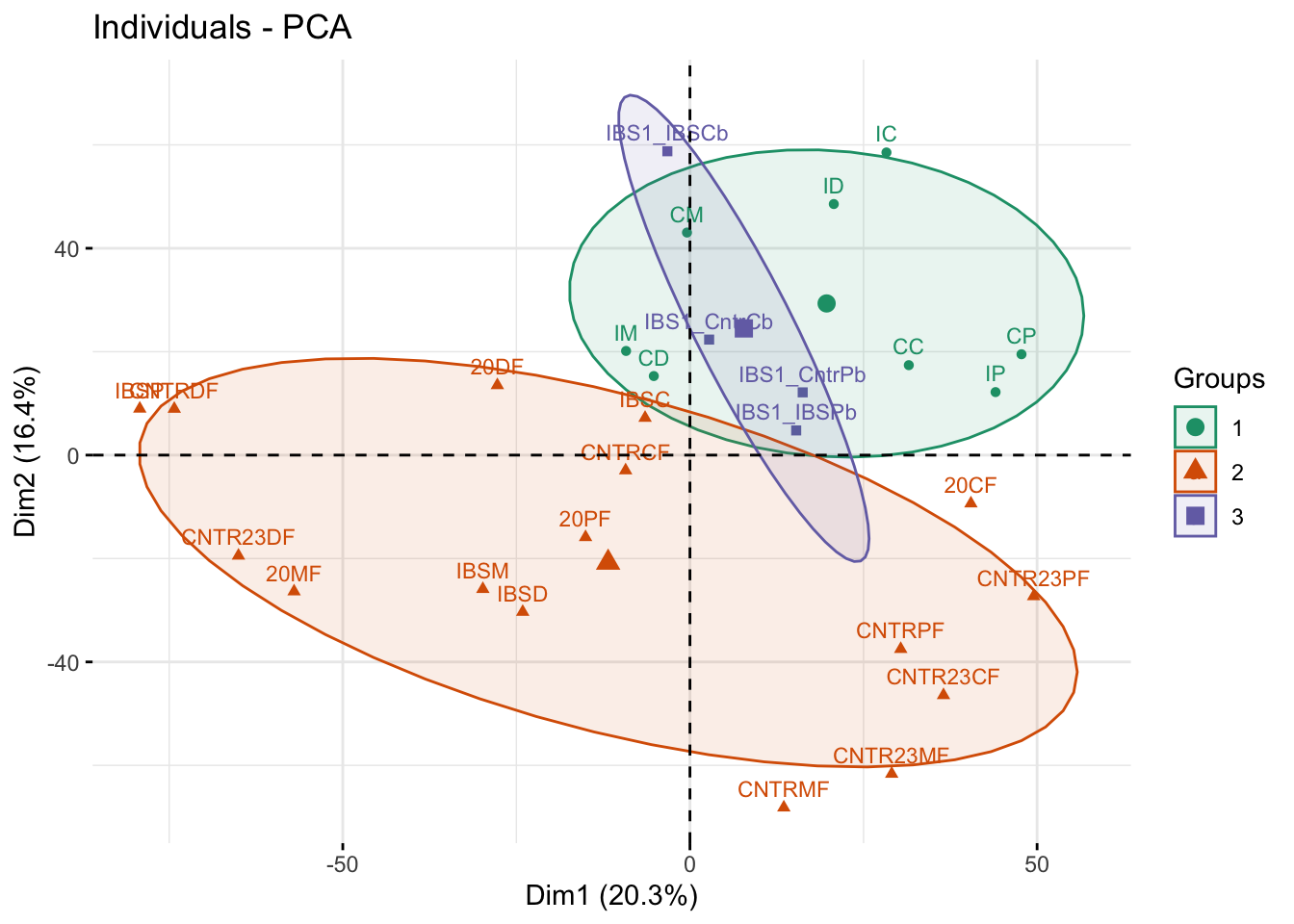
A través de esta visualización pudimos descubrir una falla en el diseño experimental original. Los dos primeros lotes mostrados en verde y marrón estaban equilibrados con respecto al IBS y las ratas sanas. Muestran niveles muy diferentes de variabilidad y coordenadas generales multivariadas. De hecho, hay dos efectos de confusión. Tanto las matrices como los protocolos fueron diferentes en esos dos días. Tuvimos que ejecutar un tercer lote de experimentos el día 3, representado en violeta, este protocolo usó desde el día 1 y las matrices desde el día 2. El tercer grupo se superpone fielmente con el lote 1, diciéndonos que el cambio en el protocolo fue responsable de la variabilidad.
Eliminar efectos de lotes
Mediante la combinación de las mediciones continuas del ensayoIBD y el número de lote suplementario como factor, el mapa de PCA ha proporcionado una herramienta de investigación invaluable. Este es un buen ejemplo del uso de puntos suplementarios. Los puntos del baricentro medio se crean utilizando los medios de grupo de los puntos en cada uno de los tres grupos y sirven como marcadores adicionales en la gráfica.
Podemos decidir realinear los tres grupos restando las medias del grupo para que todos los lotes estén centrados en el origen. Una forma un poco más eficaz es utilizar la función ComBat disponible en el paquete sva. Esta función utiliza un método similar, pero un poco más sofisticado (enfoque empírico de mezcla de Bayes (Leek et al. 2010)). Podemos ver su efecto en los datos al rehacer nuestro PCA robusto (vea el resultado en la Figura 9.14):
model0 = model.matrix(~1, day)
combatIBD = ComBat(dat = assayIBD, batch = day, mod = model0)## Found3batches## Adjusting for0covariate(s) or covariate level(s)## Standardizing Data across genes## Fitting L/S model and finding priors## Finding parametric adjustments## Adjusting the DatapcaDayBatRM = rankthreshPCA(combatIBD)
fviz(pcaDayBatRM, element = "ind", geom = c("point", "text"),
habillage = day, repel=TRUE, palette = "Dark2", addEllipses = TRUE,
ellipse.type = "convex", axes =c(1,2)) + coord_fixed() + ggtitle("")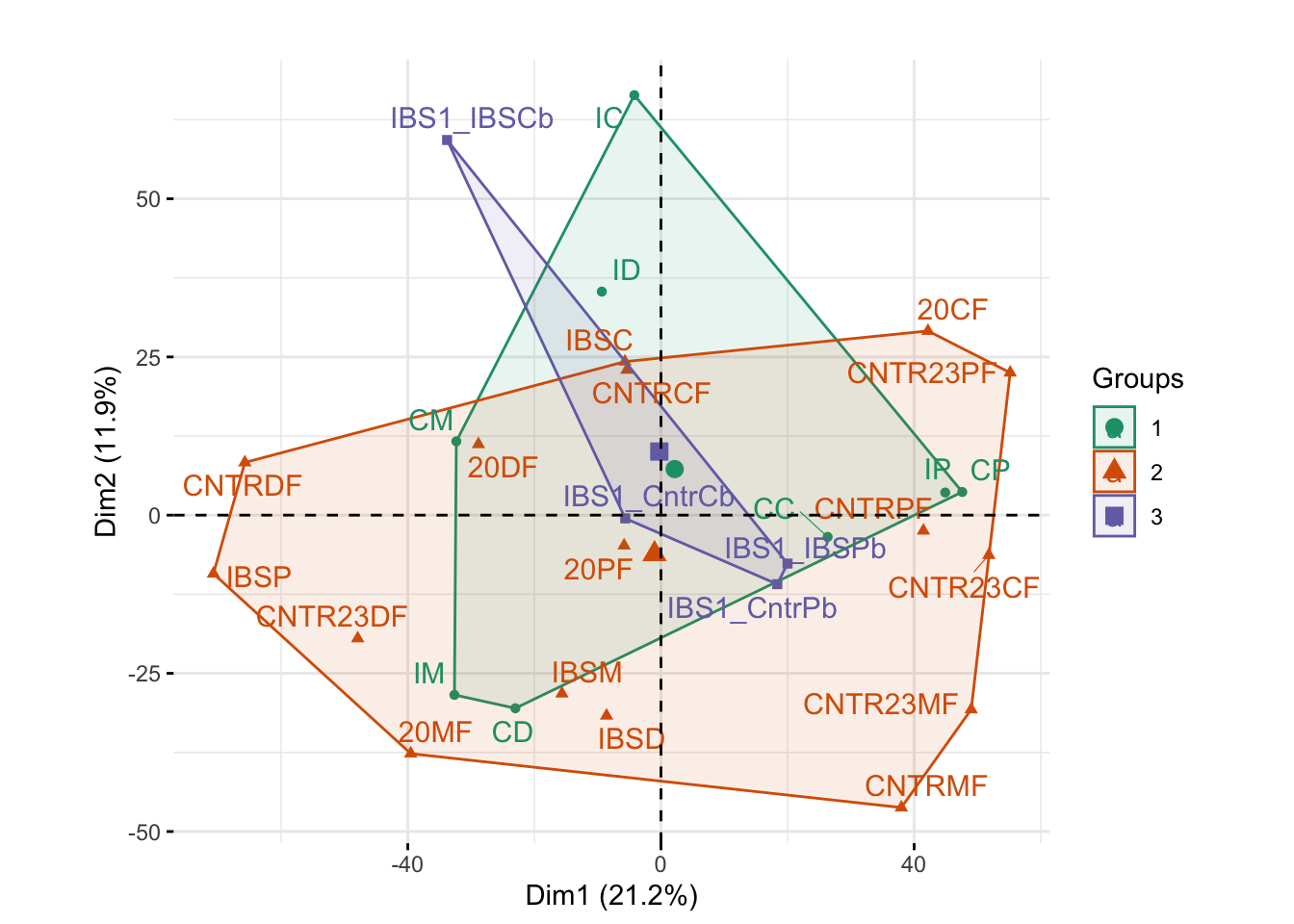
Los datos modificados con los efectos por lotes eliminados ahora muestran tres grupos de lotes muy superpuestos y centrados casi en el origen.
10.3 Análisis de correspondencia para tablas de contingencia
Tablas cruzadas y de contingencia
Los datos categóricos abundan en entornos biológicos: estado de secuencia (CpG / no CpG), fenotipos, los taxones a menudo se codifican como factores como vimos en los primeros prácticos. La tabulación cruzada de dos de estas variables nos da una tabla de contingencia; el resultado de contar la co-ocurrencia de dos fenotipos (el sexo y el daltonismo son un ejemplo). Vimos que el primer paso es mirar la independencia de las dos variables categóricas; la medida estadística estándar de independencia utiliza la distancia chi-cuadrado. Esta cantidad reemplazará la varianza que usamos para las mediciones continuas.
Las columnas y filas de la tabla tienen el mismo “estado” y no estamos en la configuración de tipo supervisado / regresión. No veremos una división de muestra / variable; como consecuencia, las filas y columnas tendrán el mismo estado y “centraremos” tanto las filas como las columnas. Esta simetría también se traducirá en nuestro uso de biplots donde ambas dimensiones aparecen en la misma gráfica.
Transforming the data to tabular form.
Si los datos se recopilan como listas largas con cada sujeto (o muestra) asociado a sus niveles de las variables categóricas, es posible que deseemos transformarlos en una tabla de contingencia. Aquí hay un ejemplo. En la tabla 10.1, las mutaciones del VIH se tabulan como variables binarias del indicador (0/1). Estos datos luego se transforman en una matriz de co-ocurrencia de mutaciones que se muestra en la tabla 10.2.
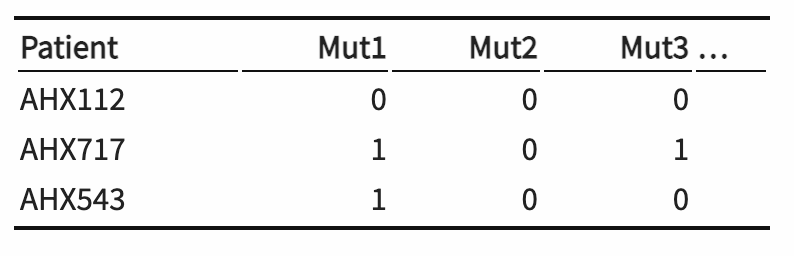
Table 10.1: Muestra por matriz de mutación..
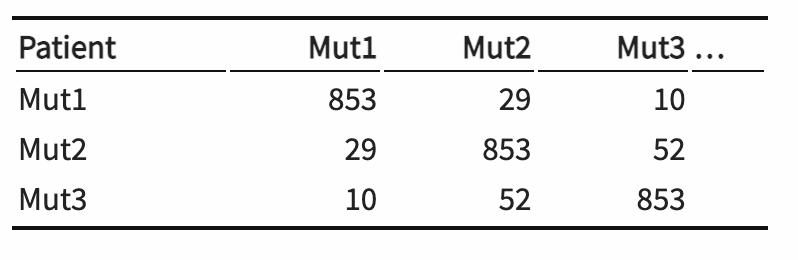
Tabla 10.2: Tabulación cruzada de las mutaciones del VIH que muestran co-ocurrencias bidireccionales.
Co-ocurrencia de color de cabello, color de ojos y fenotipo
Consideraremos una pequeña tabla para que podamos seguir el análisis en detalle. Los datos son una tabla de contingencia de co-ocurrencia fenotípica de color de cabello y color de ojos de los estudiantes como se muestra en la Tabla 10.3. Usaremos una prueba de independencia de chi cuadrado que descubre la existencia de posibles dependencias:
HairColor = HairEyeColor[,,2]
chisq.test(HairColor)## Warning in chisq.test(HairColor): Chi-squared approximation may be incorrect##
## Pearson's Chi-squared test
##
## data: HairColor
## X-squared = 106.66, df = 9, p-value < 2.2e-16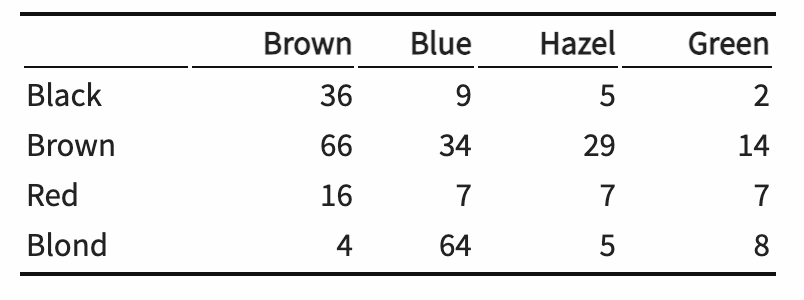
Tabla 10.3: Tabulación cruzada del color de cabello y ojos de los estudiantes
Sin embargo, no es suficiente afirmar que no hay independencia entre el color del cabello y el de los ojos. Necesitamos una explicación más detallada de dónde ocurren las dependencias: ¿qué color de cabello ocurre con más frecuencia con los ojos verdes? ¿Son independientes algunos de los niveles de las variables? De hecho, podemos estudiar la desviación de la independencia utilizando una versión ponderada especial de SVD. Este método puede entenderse como una simple extensión de PCA y MDS a tablas de contingencia.
Independencia: computacional y visualmente.
Comenzamos calculando las sumas de filas y columnas; los usamos para construir la tabla que se esperaría si los dos fenotipos fueran independientes. A esta tabla esperada la llamamos HCexp.
rowsums=as.matrix(apply(HairColor,1,sum))
rowsums## [,1]
## Black 52
## Brown 143
## Red 37
## Blond 81colsums=as.matrix(apply(HairColor,2,sum))
t(colsums)## Brown Blue Hazel Green
## [1,] 122 114 46 31HCexp=rowsums%*%t(colsums)/sum(colsums)sum((HairColor - HCexp)^2/HCexp)## [1] 106.6637Podemos estudiar estos residuos de la tabla esperada, primero numéricamente y luego en la siguiente figura.
round(t(HairColor-HCexp))## Hair
## Eye Black Brown Red Blond
## Brown 16 10 2 -28
## Blue -10 -18 -6 34
## Hazel -3 8 2 -7
## Green -3 0 3 0library("vcd")##
## Attaching package: 'vcd'## The following object is masked from 'package:GenomicRanges':
##
## tile## The following object is masked from 'package:IRanges':
##
## tilemosaicplot(HairColor,shade=TRUE,las=1,type="pearson",cex.axis=0.7,main="")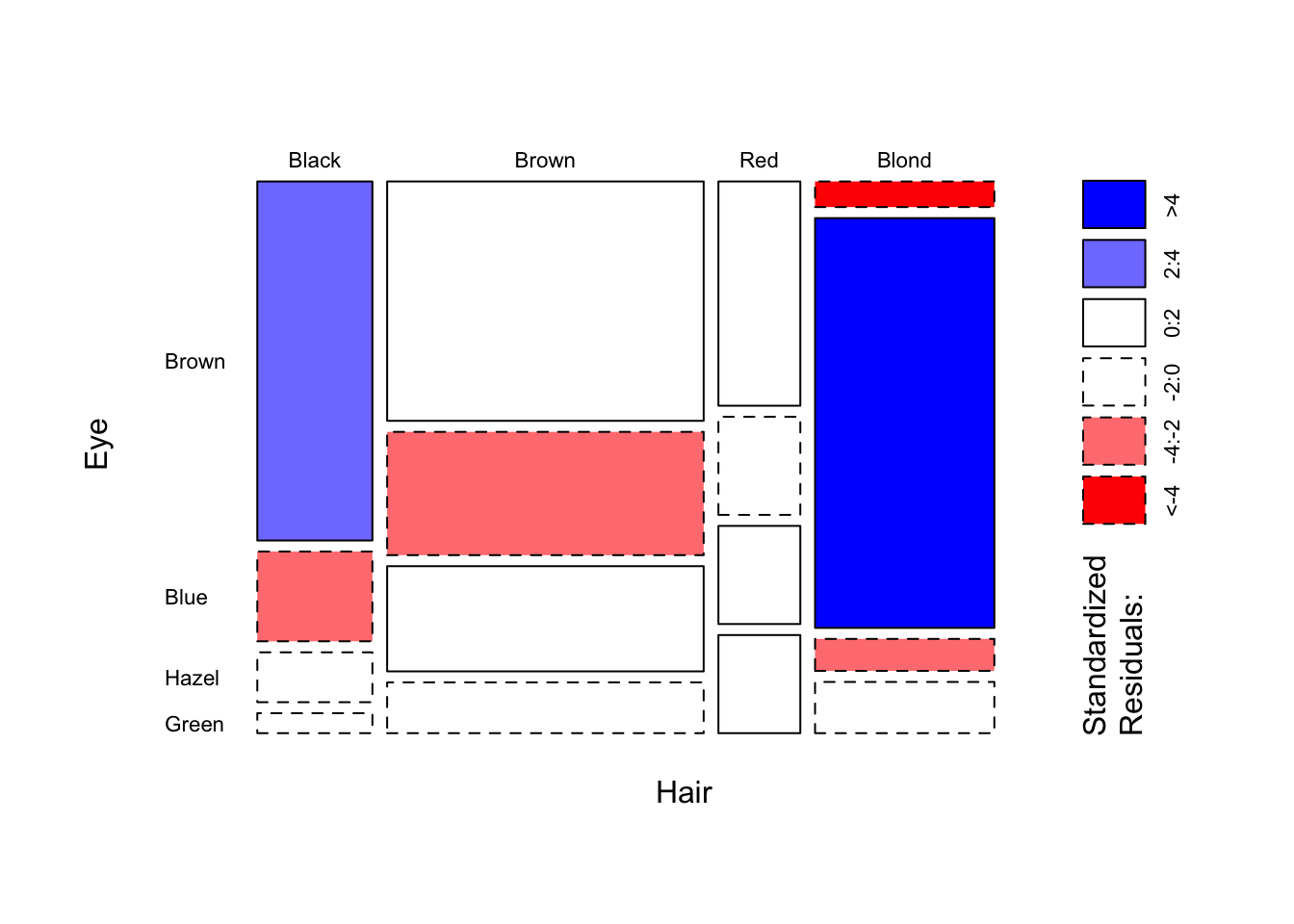 Visualización de la salida de la independencia. Ahora, las cajas son proporcionales en tamaño a los recuentos observados reales y ya no tenemos una propiedad “rectangular”. La desviación de la independencia se mide en la distancia Chi-cuadrado para cada una de las cajas y se colorea según si los residuos son grandes y positivos. El azul oscuro indica una asociación positiva, por ejemplo, entre los ojos azules y el cabello rubio, el rojo indica una asociación negativa, como en el caso del cabello rubio y los ojos marrones.
Visualización de la salida de la independencia. Ahora, las cajas son proporcionales en tamaño a los recuentos observados reales y ya no tenemos una propiedad “rectangular”. La desviación de la independencia se mide en la distancia Chi-cuadrado para cada una de las cajas y se colorea según si los residuos son grandes y positivos. El azul oscuro indica una asociación positiva, por ejemplo, entre los ojos azules y el cabello rubio, el rojo indica una asociación negativa, como en el caso del cabello rubio y los ojos marrones.
Una vez que hemos comprobado que las dos variables no son independientes, usamos una escala multidimensional ponderada usando distancias χ2 para visualizar las asociaciones.
El método se llama Análisis de correspondencia (CA) o Escalado dual y hay varios paquetes R que lo implementan.
Aquí hacemos un biplot simple de los colores de Cabello y Ojos.
HC=as.data.frame.matrix(HairColor)
coaHC=dudi.coa(HC,scannf=FALSE,nf=2)
round(coaHC$eig[1:3]/sum(coaHC$eig)*100)## [1] 89 10 2fviz_ca_biplot(coaHC,repel=TRUE,col.col="brown", col.row="purple") +
ggtitle("") + ylim(c(-0.5,0.5))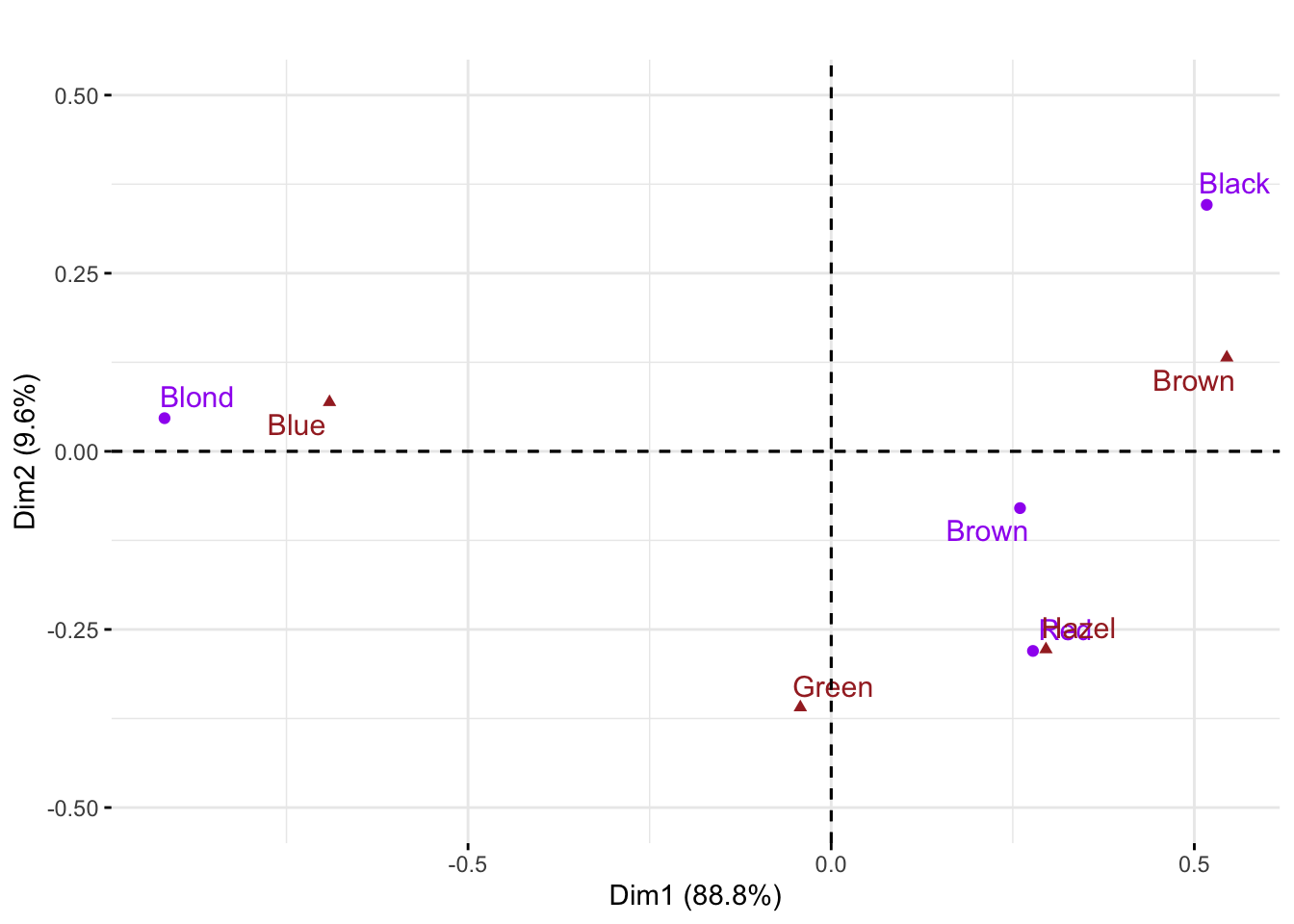
El gráfico CA da una representación de una gran proporción de la distancia chi-cuadrado entre los datos y los valores esperados bajo independencia. El primer eje muestra un contraste entre los estudiantes de cabello negro y rubio, reflejado por la oposición de ojos marrones y ojos azules. En CA las dos categorías juegan roles simétricos y podemos interpretar la proximidad de ojos azules y cabello rubio significa que hay una fuerte co-ocurrencia de estas categorías.
Interpretando el biplot CA tiene una propiedad baricéntrica especial, la escala biplot se elige de modo que los puntos de fila se coloquen en el centro de gravedad de los niveles de columna con sus respectivos pesos. Por ejemplo, el punto de la columna de ojos azules está en el centro de gravedad del (negro, marrón, rojo, rubio) con pesos proporcionales a (9,34,7,64). El punto de la fila Rubio está muy ponderado, por eso la figura anterior muestra Rubio y Azul bastante juntos.
10.4 Encontrar tiempo … y otros gradientes importantes.
Todos los métodos que hemos estudiado en las últimas secciones se conocen comúnmente como métodos de ordenación. De la misma manera que la agrupación nos permitió detectar e interpretar un factor oculto / variable categórica, la ordenación nos permite detectar e interpretar un orden oculto, gradiente o variable latente en los datos.
Los ecologistas tienen una larga historia de interpretar los arcos formados por puntos de observación en el análisis de correspondencia (CA) y componentes principales (PCA) como gradientes ecológicos (Prentice 1977). Ilustremos esto primero con un conjunto de datos muy simple en el que realizamos un análisis de correspondencia.
load("data_cap8910//lakes.RData")
lakelike[1:3,1:8]## plant1 plant2 plant3 plant4 plant5 plant6 plant7 plant8
## loc1 6 4 0 3 0 0 0 0
## loc2 4 5 5 3 4 2 0 0
## loc3 3 4 7 4 5 2 1 1reslake=dudi.coa(lakelike,scannf=FALSE,nf=2)
round(reslake$eig[1:8]/sum(reslake$eig),2)## [1] 0.56 0.25 0.09 0.03 0.03 0.02 0.01 0.00fviz_ca_row(reslake,repel=TRUE)+ggtitle("")+ylim(c(-0.55,1.7))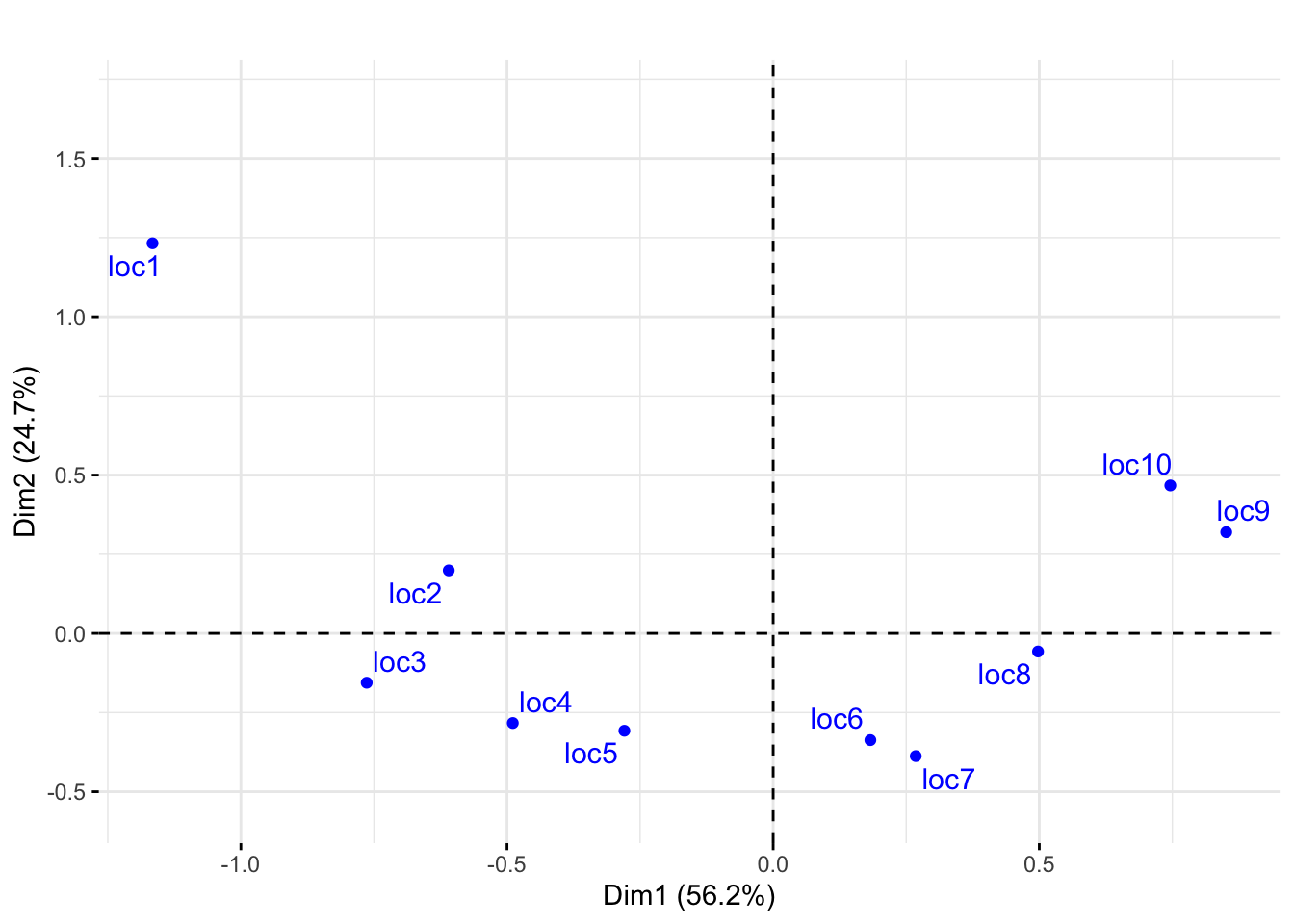
fviz_ca_biplot(reslake,repel=TRUE)+ggtitle("")+ylim(c(-0.55,1.7))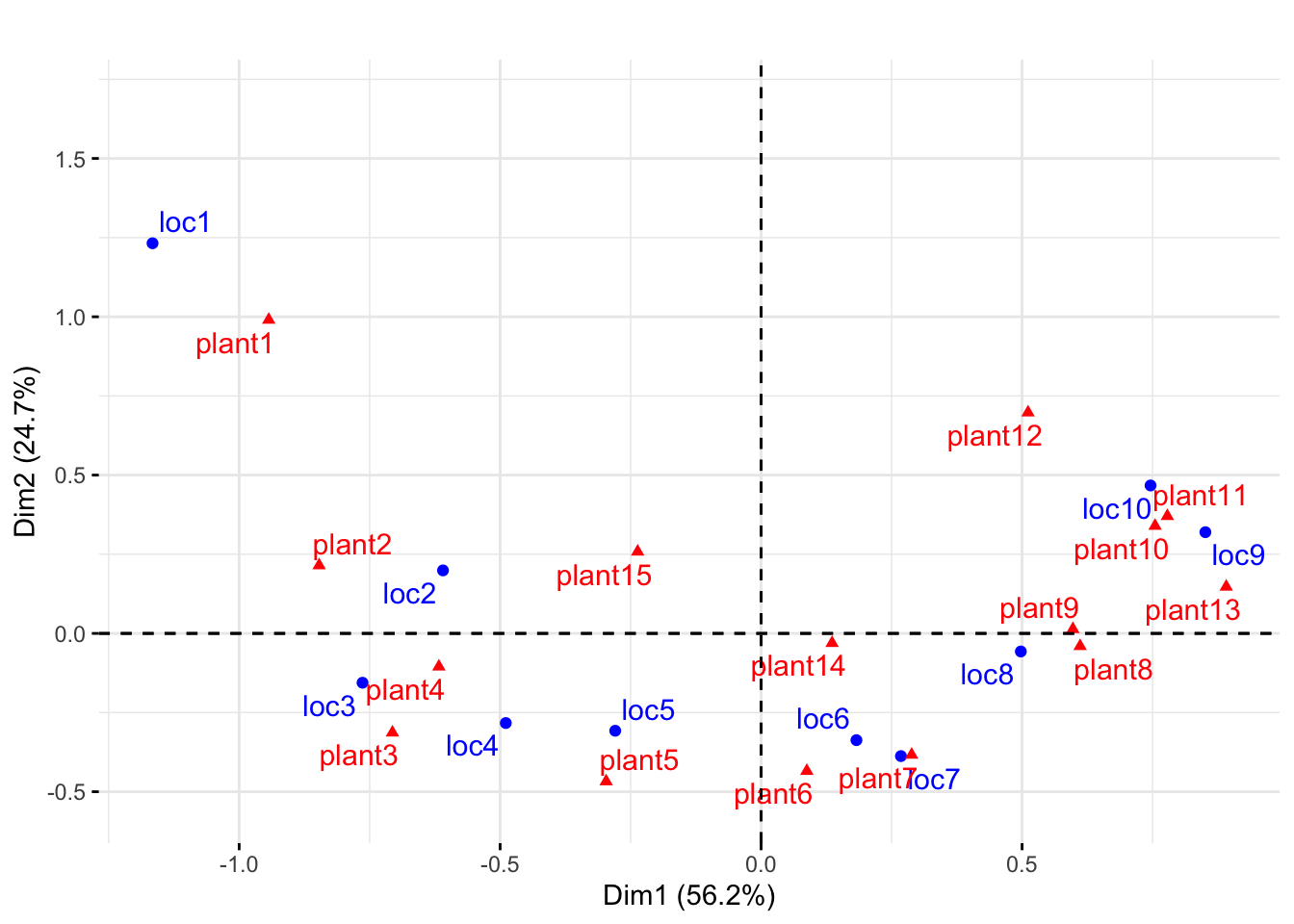
Las ubicaciones cercanas al lago están ordenadas a lo largo de un arco como se muestra en la gráfica superior. En el biplot de abajo, podemos ver qué plantas son más frecuentes en qué ubicaciones mirando los triángulos rojos más cercanos a los puntos azules.
Graficamos tanto los puntos de ubicación de las filas (en el gráfico superior de la figura) como el biplot de la ubicación y las especies de plantas en el par inferior de la figura.
10.4.1 Dinámica del desarrollo celular.
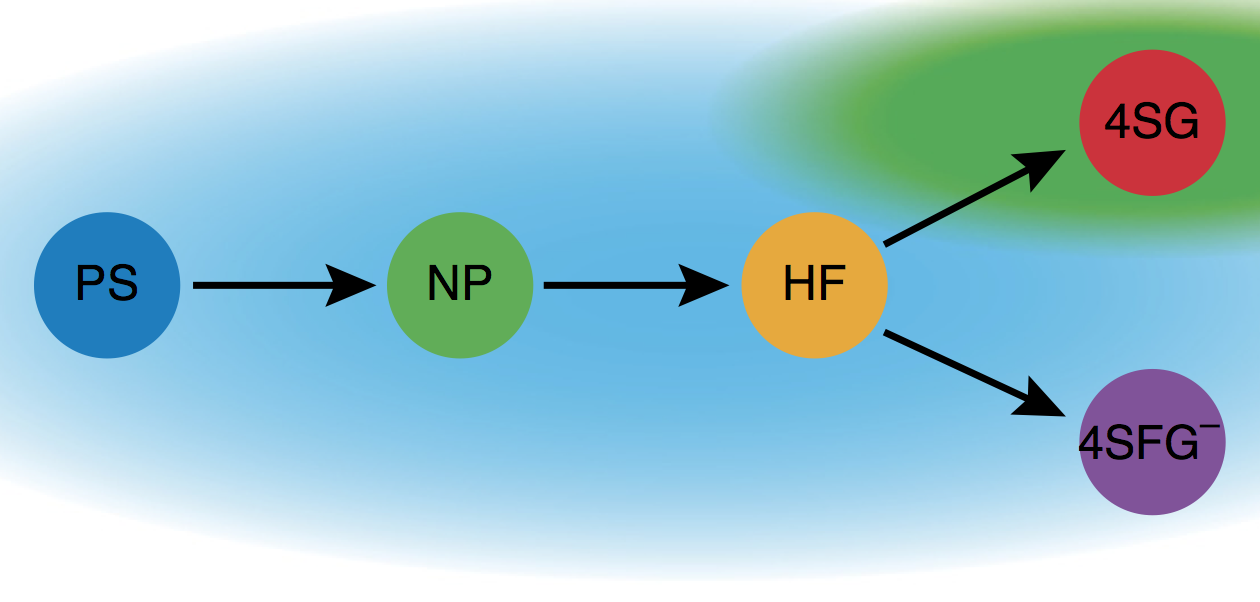
Ahora analizaremos un conjunto de datos más interesante que fue publicado por Moignard et al. (2015). Este artículo describe la dinámica del desarrollo de las células sanguíneas. Los datos son mediciones de expresión génica de células individuales de 3.934 células con potencial sanguíneo y endotelial de cinco poblaciones entre los días embrionarios E7.0 y E8.25.
Recuerdar que hay varias distancias diferentes disponibles para comparar nuestras celdas. Aquí, comenzamos calculando tanto una distancia \(L^2\) como la distancia \(ℓ_1\) entre las 3934 celdas.
library(SummarizedExperiment)
Moignard = readRDS("data_cap8910//Moignard.rds")
cellt = rowData(Moignard)$celltypes
colsn = c("red", "purple", "orange", "green", "blue")
blom = assay(Moignard)
dist2n.euclid = dist(blom)
dist1n.l1 = dist(blom, "manhattan")El escalado multidimensional clásico en estas dos matrices de distancias se puede realizar utilizando:
ce1Mds = cmdscale(dist1n.l1, k = 20, eig = TRUE)
ce2Mds = cmdscale(dist2n.euclid, k = 20, eig = TRUE)
perc1 = round(100*sum(ce1Mds$eig[1:2])/sum(ce1Mds$eig))
perc2 = round(100*sum(ce2Mds$eig[1:2])/sum(ce2Mds$eig))Observamos la dimensión subyacente y vemos en la siguiente figura que dos dimensiones pueden proporcionar una fracción sustancial de la varianza.
plotbar(ce1Mds, m = 4)
`plotbar(ce2Mds, m = 4)

Las primeras 2 coordenadas explican el 78% de la variabilidad cuando se usa la distancia \(ℓ_1\) entre celdas y el 57% cuando se usa la distancia \(L^2\). Vemos en la siguiente figura (A) el primer plano para el MDS en las distancias ℓ1 entre celdas:
c1mds = as_tibble(ce1Mds$points[, 1:2]) %>%
setNames(paste0("L1_PCo", 1:2))## Warning: The `x` argument of `as_tibble.matrix()` must have unique column names if `.name_repair` is omitted as of tibble 2.0.0.
## Using compatibility `.name_repair`.ggplot(c1mds,aes(x = L1_PCo1,y = L1_PCo2, color = cellt)) +
geom_point(aes(color = cellt), alpha = 0.6) +
scale_colour_manual(values = colsn) + guides(color = FALSE)## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.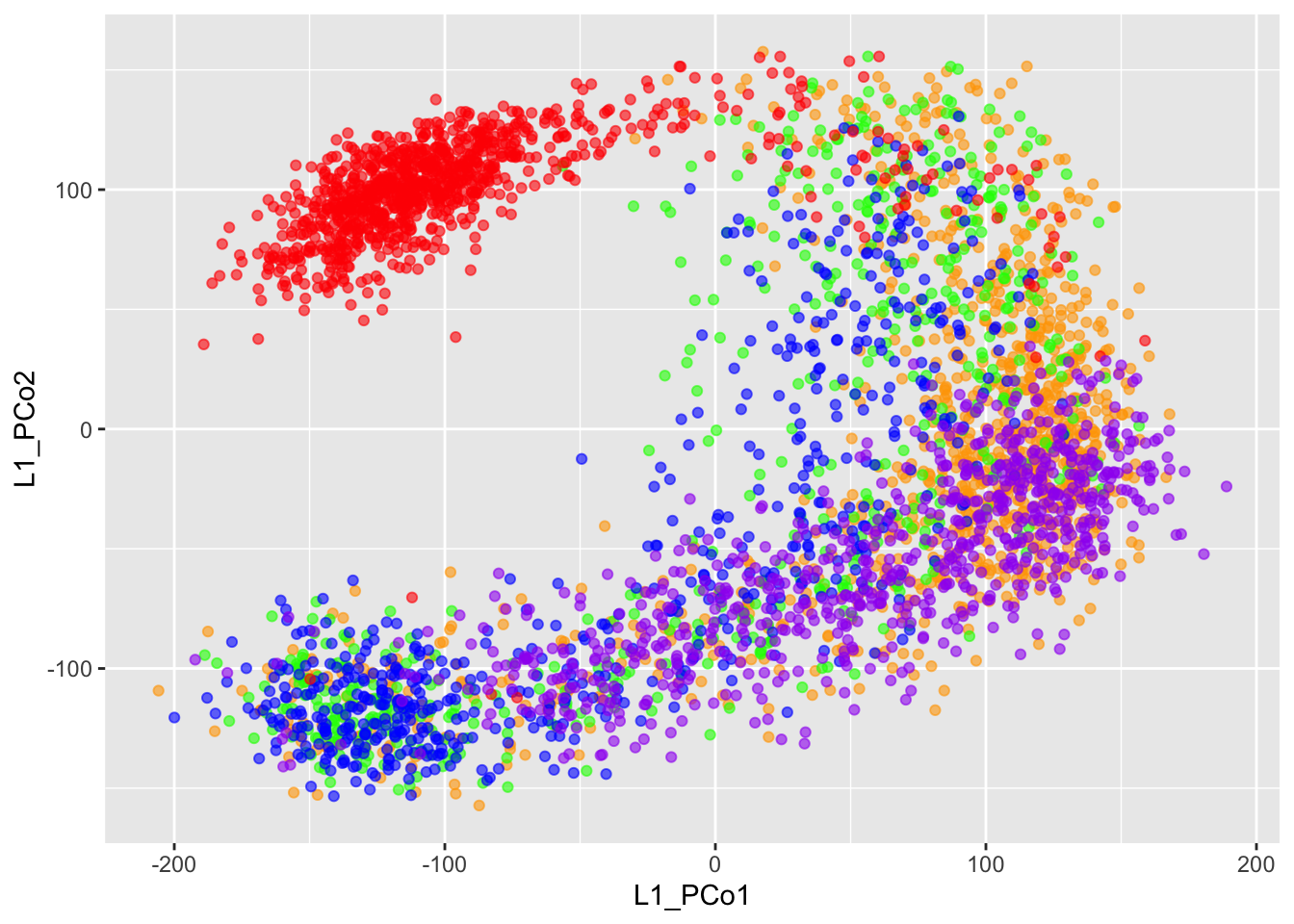
c2mds = as_tibble(ce1Mds$points[, 1:2]) %>%
setNames(paste0("L2_PCo", 1:2))
ggplot(c2mds,aes(x=L2_PCo1,y=L2_PCo2, color = cellt)) +
geom_point(aes(color = cellt), alpha = 0.6) +
scale_colour_manual(values = colsn) + guides(color = FALSE)## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.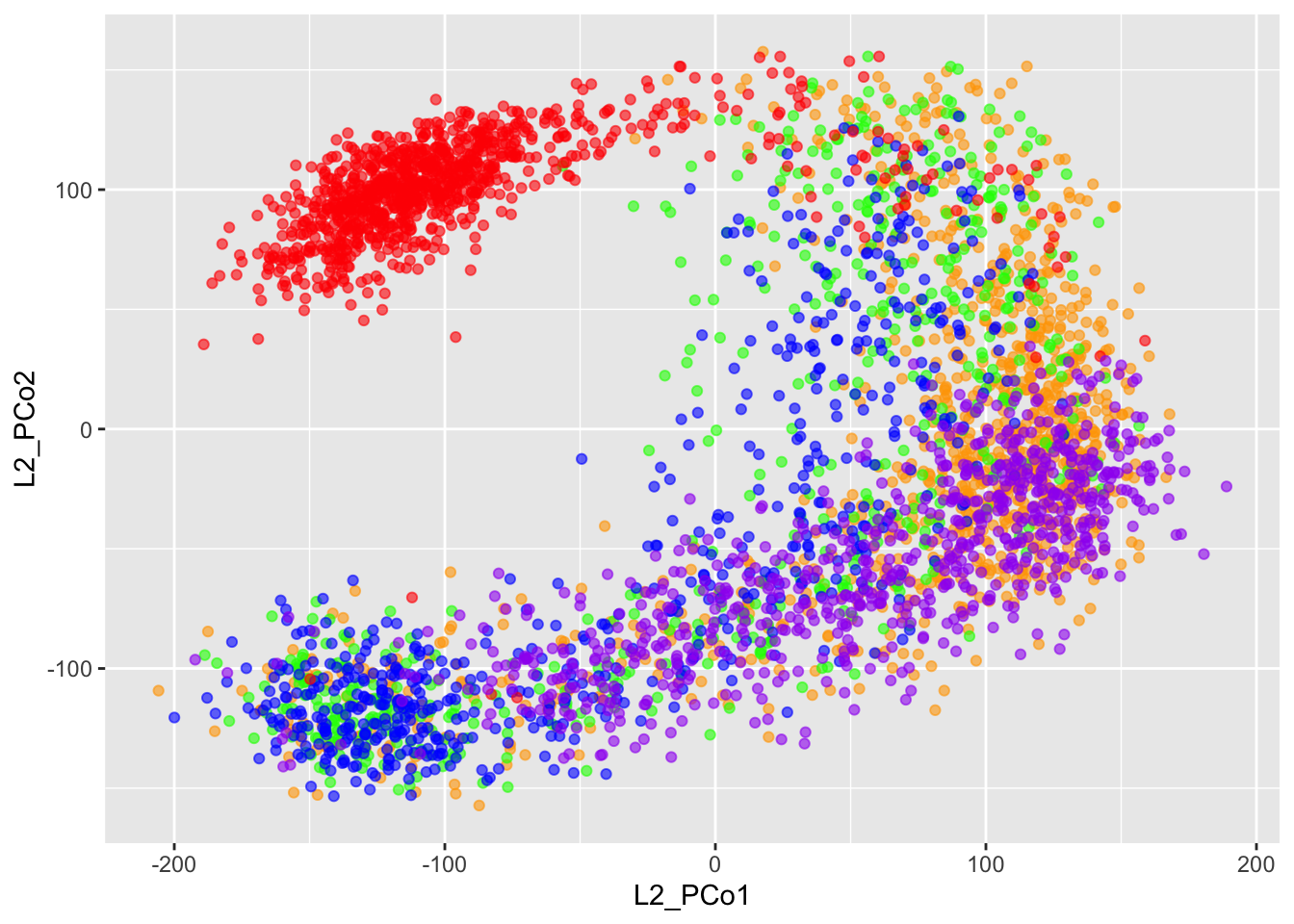 La figura B se crea de la misma manera y muestra la proyección bidimensional creada usando MDS en las distancias \(L^2\).
La figura B se crea de la misma manera y muestra la proyección bidimensional creada usando MDS en las distancias \(L^2\).
Las figuras muestran que ambas distancias (\(ℓ_1\) y \(L^2\)) dan el mismo primer plano para el MDS con representaciones muy similares del gradiente subyacente seguido de las celdas.
Podemos ver en además, que las celdas no están distribuidas uniformemente en las dimensiones inferiores que hemos estado viendo, vemos una organización definida de los puntos. Todas las células de tipo 4SG representadas en rojo forman un grupo alargado que está mucho menos mezclado con los otros tipos de células.
10.5 Técnicas de MultiTable
Los estudios actuales a menudo intentan cuantificar la variación en las mediciones microbianas, genómicas y metabólicas en diferentes condiciones experimentales. Como resultado, es común realizar múltiples ensayos en las mismas muestras biológicas y preguntar qué características (microorganismos, genes o metabolitos, por ejemplo) están asociadas con diferentes condiciones de muestra. Hay muchas formas de abordar estas cuestiones. Cuál aplicar depende del enfoque del estudio.
En el capítulo 9 de el libro Modern Statistic for Modern Biology se revisan en detalle las técnicas:
- Covariación, inercia, coeficiente de co-inercia y RV
- Coeficiente de Mantel y una prueba de correlación de distancia
- Análisis de correlación canónica (CCA)
- Análisis de correlación canónica dispersa (sCCA)
- Análisis de correspondencia canónico (o restringido) (CCpnA)
Aquí revisaremos un ejemplo de Análisis de correlación canónica dispersa (sCCA) a modo representativo. Para ver en detalle las otras técnicas revisar el capítulo del libro mencionado.
10.5.1 Análisis de correlación canónica dispersa (sCCA)
Cuando el número de variables en cada tabla es muy grande, encontrar dos vectores muy correlacionados puede ser demasiado fácil e inestable: tenemos demasiados grados de libertad.
Entonces es beneficioso agregar una penalización que mantiene el número de coeficientes distintos de cero al mínimo. Este enfoque se denomina análisis de correlación canónica dispersa (CCA dispersa o sCCA), un método muy adecuado tanto para comparaciones exploratorias entre muestras como para la identificación de características con covariación interesante. Usaremos una implementación del paquete PMA.
Aquí estudiamos un conjunto de datos recopilados por Kashyap et al. (2013) con dos tablas. Una es una tabla de contingencia de abundancias bacterianas y otra una tabla de abundancia de metabolitos. Hay 12 muestras, entonces n = 12. La tabla de metabolitos tiene mediciones en la característica p = 637 y las abundancias bacterianas tenían un total de \(q = 20.609\) OTUs, que filtraremos hasta alrededor de 200. Comenzamos cargando los datos.
mb_path = "data_cap8910//metabolites.csv"
library("genefilter")
load("data_cap8910//microbe.rda")
metab = read.csv(mb_path, row.names = 1) %>% as.matrixPrimero filtramos hacia las bacterias y metabolitos de interés, eliminando (“a mano”) aquellos que son cero en muchas muestras y dando un umbral superior de 50 a los valores grandes. Transformamos los datos para debilitar las colas pesadas.
library(phyloseq)
metab = metab[rowSums(metab == 0) <= 3, ]
microbe = prune_taxa(taxa_sums(microbe) > 4, microbe)## Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'## Also defined by 'tidytree'microbe = filter_taxa(microbe, filterfun(kOverA(3, 2)), TRUE)## Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
## Also defined by 'tidytree'metab = log(1 + metab, base = 10)
X = as.matrix(otu_table(microbe))
X = log(1 + X, base=10)Un segundo paso en nuestro análisis preliminar es ver si existe alguna asociación entre las dos matrices usando el RV.test del paquete ade4:
library(ade4)
colnames(metab)=colnames(X)
pca1 = dudi.pca(t(metab), scal = TRUE, scann = FALSE)
pca2 = dudi.pca(t(X), scal = TRUE, scann = FALSE)
rv1 = RV.rtest(pca1$tab, pca2$tab, 999)
rv1## Monte-Carlo test
## Call: RV.rtest(df1 = pca1$tab, df2 = pca2$tab, nrepet = 999)
##
## Observation: 0.8400429
##
## Based on 999 replicates
## Simulated p-value: 0.004
## Alternative hypothesis: greater
##
## Std.Obs Expectation Variance
## 5.549626024 0.315495787 0.008933911Ahora podemos aplicar CCA. Este método compara conjuntos de características en tablas de datos de alta dimensión, donde puede haber más características medidas que muestras. En el proceso, elige un subconjunto de características disponibles que capturan la mayor covarianza; estas son las características que reflejan las señales presentes en varias tablas. Luego aplicamos PCA a este subconjunto seleccionado de características. En este sentido, usamos CCA como un procedimiento de selección, más que como un método de ordenación.
La implementación está a continuación. Los parámetros penaltyx y penaltyz son penalizaciones por escasez. Los valores más pequeños de penaltyx darán como resultado menos microorganismos seleccionados, de manera similar penaltyz modula el número de metabolitos seleccionados. Los ajustamos manualmente para facilitar la interpretación posterior; por lo general, preferimos más escasez de la que proporcionarían los parámetros predeterminados.
library("PMA")
ccaRes = CCA(t(X), t(metab), penaltyx = 0.15, penaltyz = 0.15,
typex = "standard", typez = "standard")## 123456789ccaRes## Call: CCA(x = t(X), z = t(metab), typex = "standard", typez = "standard",
## penaltyx = 0.15, penaltyz = 0.15)
##
##
## Num non-zeros u's: 5
## Num non-zeros v's: 16
## Type of x: standard
## Type of z: standard
## Penalty for x: L1 bound is 0.15
## Penalty for z: L1 bound is 0.15
## Cor(Xu,Zv): 0.9904707Con estos parámetros, se seleccionaron 5 bacterias y 16 metabolitos en función de su capacidad para explicar la covariación entre tablas. Además, estas características dan como resultado una correlación de 0,99 entre las dos tablas. Interpretamos que esto significa que los datos microbianos y metabolómicos reflejan señales subyacentes similares, y que estas señales pueden aproximarse bien por las características seleccionadas. Sin embargo, tener cuidado con el valor de correlación, ya que las puntuaciones están lejos de la nube normal bivariada habitual. Además, tenga en cuenta que es posible que otros subconjuntos de características puedan explicar los datos igualmente bien: el CCA ha minimizado la redundancia entre las características, pero no garantiza que estas sean las características “verdaderas” en ningún sentido.
No obstante, todavía podemos usar estas 21 funciones para comprimir la información de las dos tablas sin mucha pérdida. Para relacionar los metabolitos recuperados y las OTUs con las características de las muestras en las que se midieron, los usamos como entrada para un PCA ordinario. Hemos omitido el código que usamos para generar la figura a continuación, remitimos al lector al material en línea que acompaña al libro o al flujo de trabajo publicado en Ben J Callahan et al. (2016).
La figura muestra el triplot de PCA, donde mostramos diferentes tipos de muestras y las características multidominio (metabolitos y OTU). Esto permite la comparación entre las muestras medidas (triángulos para knockout y círculos para wild type) y caracteriza la influencia de las diferentes características: diamantes con etiquetas de texto. Por ejemplo, vemos que la principal variación en los datos se da entre las muestras de PD y ST, que corresponden a las diferentes dietas. Además, los valores grandes de 15 de las características están asociados con el estado ST, mientras que los valores pequeños de 5 de ellos indican el estado de PD.

Un triplot de PCA producido a partir de características seleccionadas de CCA de múltiples tipos de datos (metabolitos y OTU).
La ventaja del screening CCA ahora es clara: podemos mostrar la mayor parte de la variación entre las muestras utilizando un gráfico relativamente simple y podemos evitar graficar los cientos de puntos adicionales que serían necesarios para mostrar todas las características.