Introducción a phyloseq y a análisis de diversidad
ISME Latin America, Septiembre 2019
0.1 Acerca del curso
El tutorial a continuación fue creado especialmente para guiar el trabajo práctico del curso pre-congreso ISME Latin America 2019: Análisis de datos bioinformáticos para metagenomas y amplicones usando R. A realizarse el próximo 9 y 10 de septiembre en la Universidad Técnica Federico Santa María, Valparaíso, Chile.
 Ir a la página de inicio del curso
Ir a la página de inicio del curso
El curso cuenta con 6 unidades. Ahora, usted se encuentra en la unidad: Introducción a phyloseq y a análisis de diversidad 
Otras unidades del curso son:
Introducción a R: Manipulación de datos y visualización
Análisis de secuencias de 16S con DADA2
Búsqueda de genes de interés en datos de metagenómica shotgun
Visualización y curación de genomas ensamblados desde metagenomas (MAGs)




Profesor: Dr. Eduardo Castro-Nallar (eduardo.castro@unab.cl)
- Ayudantes:
- Dr. Florence Gutzwiller (florence.gutzwiller@gmail.com)
- M.Sc. Katterinne N. Mendez (mendez.katterinne@gmail.com)
Centro de Bioinformática y Biología Integrativa | Universidad Andrés Bello

1 Instalación de paquetes de R
Los siguientes 3 scripts te mostrarán una manera eficiente de instalar y cargar la lista de paquetes según su repositorio de origen, ya que cada repositorio tiene su propia función para instlar sus paquetes.
- Primero, enlistar los paquetes necesarios en diferentes vectores dependiendo de su repositorio de origen.
# Definir paquetes
## Repositorio CRAN
cran_packages <- c("bookdown", "knitr", "tidyverse", "plyr", "grid", "gridExtra", "kableExtra", "xtable", "ggpubr")
## Repositorio Bioconductor
bioc_packages <- c("phyloseq", "dada2", "DECIPHER", "phangorn", "ggpubr", "BiocManager","DESeq2", "microbiome", "philr")
## Repositorio GitHub
git_source <- c("twbattaglia/btools", "gmteunisse/Fantaxtic", "MadsAlbertsen/ampvis2", "opisthokonta/tsnemicrobiota") # fuente/nombre del paquete
git_packages <- c("btools", "fantaxtic", "ampvis2", "tsnemicrobiota") # nombre del paquete- Segundo, instalar los paquetes definidos arriba usando la función correspondiente a cada repositorio.
# Instalar paquetes CRAN
.inst <- cran_packages %in% installed.packages()
if(any(!.inst)) {
install.packages(cran_packages[!.inst])
}
# Intalar paquetes BioConductor
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
.inst <- bioc_packages %in% installed.packages()
if(any(!.inst)) {
BiocManager::install(bioc_packages[!.inst])
}
# Instalar paquetes GitHub
.inst <- git_source %in% installed.packages()
if(any(!.inst)) {
devtools::install_github(git_source[!.inst])
}- Tercero, cargar los paquetes requeridos a la sesión actual de R.
# Cargar paquetes
sapply(c(cran_packages, bioc_packages, git_packages), require, character.only = TRUE)El paso de instalación de paquetes es necesario solamente una vez. Excepto si se quiere actualizar la versión del paquete, o bien, R ha sido desinstalado e instalado nuevamente o actualizado su versión.
Si ya tienes los paquetes instalados en tu computadora, sólo necesitas enlistar (1er script) y cargar (3er script) los paquetes. También, puedes cargarlos a la sesión actual de R usando la función library(), así:
# Cargar paquetes
library(tidyverse)
library(plyr)
library(grid)
library(gridExtra)
library(kableExtr)
library(xtable)
library(ggpubr)
library(phyloseq)
library(dada2)
library(DECIPHER)
library(phangorn)
library(ggpubr)
library(BiocManager)
library(DESeq2)
library(microbiome)
library(philr)
library(btools)
library(fantaxtic)
library(ampvis2)
library(tsnemicrobiota)2 Introducción a phyloseq
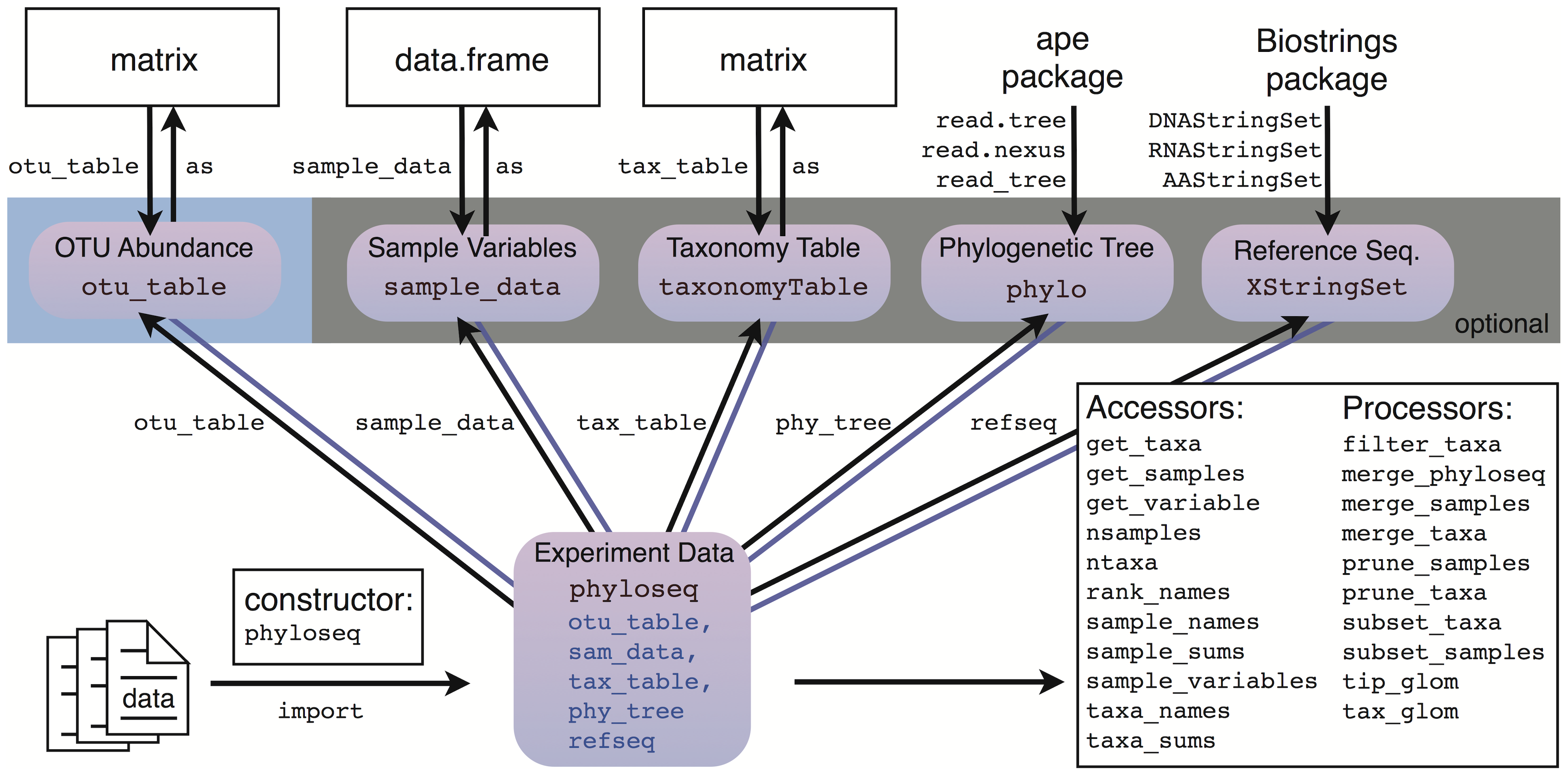
Phyloseq es un paquete de Bioconductor (Open Source Software For Bioinformatics) para la manipulación y análisis de datos metagenómicos generados por metodologías de secuenciación de alto rendimiento. phyloseq es una herramienta para importar, guardar, analizar y visualizar éste tipo de datos después de haber sido procesados inicialmente, e.g., ensamblaje de novo, ASVs u OTUs (clustered), incluyendo otros importantes datos asociados (si están disponibles): tabla de observaciones asociadas a cada muestra (e.g., especie, localización geográfica, temperatura, etc.), conocida como sample data o metadata, árbol filogenético, e identificación taxonómica de cada OTU. La estructura del paquete phyloseq consiste en una serie de funciones de acceso y de proceso de objetos phyloseq. Estos objetos están compuestos de cuatro componentes que almacenan las cuentas de reads, la metadata, la taxonomía y el árbol filogenético. El paquete también provee una serie de herramientas para importar datos de otros programas. El siguiente diagrama muestra la estructura completa de phyloseq.
Si no tienes el objeto phyloseq
psgenerado por DADA2 puedes descargarlo haciendo clic: AQUÍEn la terminal de R cargamos el objeto phyloseq:
# Leémos el objeto phyloseq del análisis por DADA2
readRDS("ps.RDS") -> psd
psd## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1476 taxa and 98 samples ]
## sample_data() Sample Data: [ 98 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 1476 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1476 tips and 1474 internal nodes ]2.1 Control de calidad del análisis de 16S
Lo primero que podemos mirar es la prevalencia de las features taxonómicas.
- Primero creamos un data frame con los valores de prevalencia, luego les agregamos la taxonomía y graficamos.
# Computamos prevalencia para cada feature y la guardamos en un data frame
prevdf = apply(X = otu_table(psd),
MARGIN = ifelse(taxa_are_rows(psd), yes = 1, no = 2),
FUN = function(x){sum(x > 0)})
# Le agregamos la taxonomía
prevdf = data.frame(Prevalence = prevdf,
TotalAbundance = taxa_sums(psd),
tax_table(psd))
plyr::ddply(prevdf, "Phylum", function(df1){cbind(mean(df1$Prevalence),sum(df1$Prevalence))}) -> dfprev
kable(dfprev)| Phylum | 1 | 2 |
|---|---|---|
| Acidobacteria | 3.800000 | 38 |
| Actinobacteria | 5.041667 | 363 |
| Bacteroidetes | 5.719388 | 2242 |
| BRC1 | 1.000000 | 1 |
| Chloroflexi | 1.750000 | 21 |
| Ciliophora | 3.000000 | 9 |
| Cyanobacteria | 4.223881 | 283 |
| Deinococcus-Thermus | 1.333333 | 4 |
| Epsilonbacteraeota | 2.760000 | 69 |
| Euryarchaeota | 3.714286 | 26 |
| Firmicutes | 4.685315 | 670 |
| Fusobacteria | 3.500000 | 70 |
| Gemmatimonadetes | 1.000000 | 2 |
| Kiritimatiellaeota | 1.000000 | 1 |
| Lentisphaerae | 1.250000 | 5 |
| Marinimicrobia_(SAR406_clade) | 3.500000 | 7 |
| Nanoarchaeaeota | 1.000000 | 1 |
| Nitrospinae | 1.000000 | 1 |
| Ochrophyta | 1.000000 | 1 |
| Patescibacteria | 5.473684 | 312 |
| Planctomycetes | 3.235294 | 110 |
| Proteobacteria | 8.233840 | 4331 |
| Schekmanbacteria | 1.000000 | 1 |
| Spirochaetes | 1.500000 | 3 |
| Tenericutes | 6.083333 | 73 |
| Thaumarchaeota | 4.000000 | 12 |
| Verrucomicrobia | 4.814815 | 130 |
| NA | 6.425532 | 302 |
Al examinar la tabla, es evidente que algunos Phylum aunque presentes, están muy poco representados. La columna 1 representa la media de read counts para ese Phylum, mientras que la columna 2 representa la suma. Por ejemplo, grupos como BRC1, Kiritimatiellaeota, y Nanoarchaeaeota están representados solamente por 1 read. Es muy riesgoso mantener estos grupos taxonómicos en el análisis ya que pueden corresponder a falsos positivos.
- Para filtrarlos, generamos un vector con todas las taxa que queremos filtrar.
# Definimos taxa a filtrar
filterPhyla = c("BRC1", "Deinococcus-Thermus", "Gemmatimonadetes", "Kiritimatiellaeota", "Nanoarchaeaeota", "Ochrophyta", "Schekmanbacteria", "Ciliophora", "Spirochaetes", NA)
# Procedemos a filtrar
(psd1 = subset_taxa(psd, !Phylum %in% filterPhyla))## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1414 taxa and 98 samples ]
## sample_data() Sample Data: [ 98 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 1414 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1414 tips and 1412 internal nodes ]# Además aprovechamos a remover taxa que no corresponde a microorganismos como cloroplastos, mitocondrias y otros
filterPhyla2 <- c("Chloroplast", "Mitochondria", "Eukaryota")
psd1 <- subset_taxa(psd1, !Kingdom %in% filterPhyla2)
psd1 <- subset_taxa(psd1, !Phylum %in% filterPhyla2)
psd1 <- subset_taxa(psd1, !Class %in% filterPhyla2)
psd1 <- subset_taxa(psd1, !Order %in% filterPhyla2)
psd1 <- subset_taxa(psd1, !Family %in% filterPhyla2)
psd1 <- subset_taxa(psd1, !Genus %in% filterPhyla2)- Además del filtrado que acabamos de realizar, existen otros tipos de filtrado que tienen que ver con la media de read counts por taxa, con la distribución de éstas, y con filtrar muestras bajo un número mínimo de reads.
# Filtramos taxa de acuerdo a un umbral de número medio de _read counts_, en este caso 1e-5
psd2 <- filter_taxa(psd1, function(x) mean(x) > 1e-5, TRUE)
# También podemos remover taxa que no se observe más de X veces en al menos 10% de las muestras
psd3 <- filter_taxa(psd2, function(x) sum(x > 2) > (0.1*length(x)), TRUE)
# Y finalmente filtrar muestras con menos de 1000 reads
psd4 = prune_samples(sample_sums(psd3) > 1000, psd3)
psd4## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 136 taxa and 87 samples ]
## sample_data() Sample Data: [ 87 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 136 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 136 tips and 135 internal nodes ]- Otra forma de filtrar taxa de baja prevalencia es estableciendo un umbral y luego visulizar el efecto de manera grafica.
# Seleccionamos las taxa de interés
prevdf1 = subset(prevdf, Phylum %in% get_taxa_unique(psd4, "Phylum"))
ggplot(prevdf1, aes(TotalAbundance, Prevalence / nsamples(psd),color=Phylum)) +
# Agregamos una línea para nuestro umbral
geom_hline(yintercept = 0.05, alpha = 0.5, linetype = 2) + geom_point(size = 2, alpha = 0.7) +
scale_x_log10() + xlab("Total Abundance") + ylab("Prevalence [Frac. Samples]") +
facet_wrap(~Phylum) + theme(legend.position="none")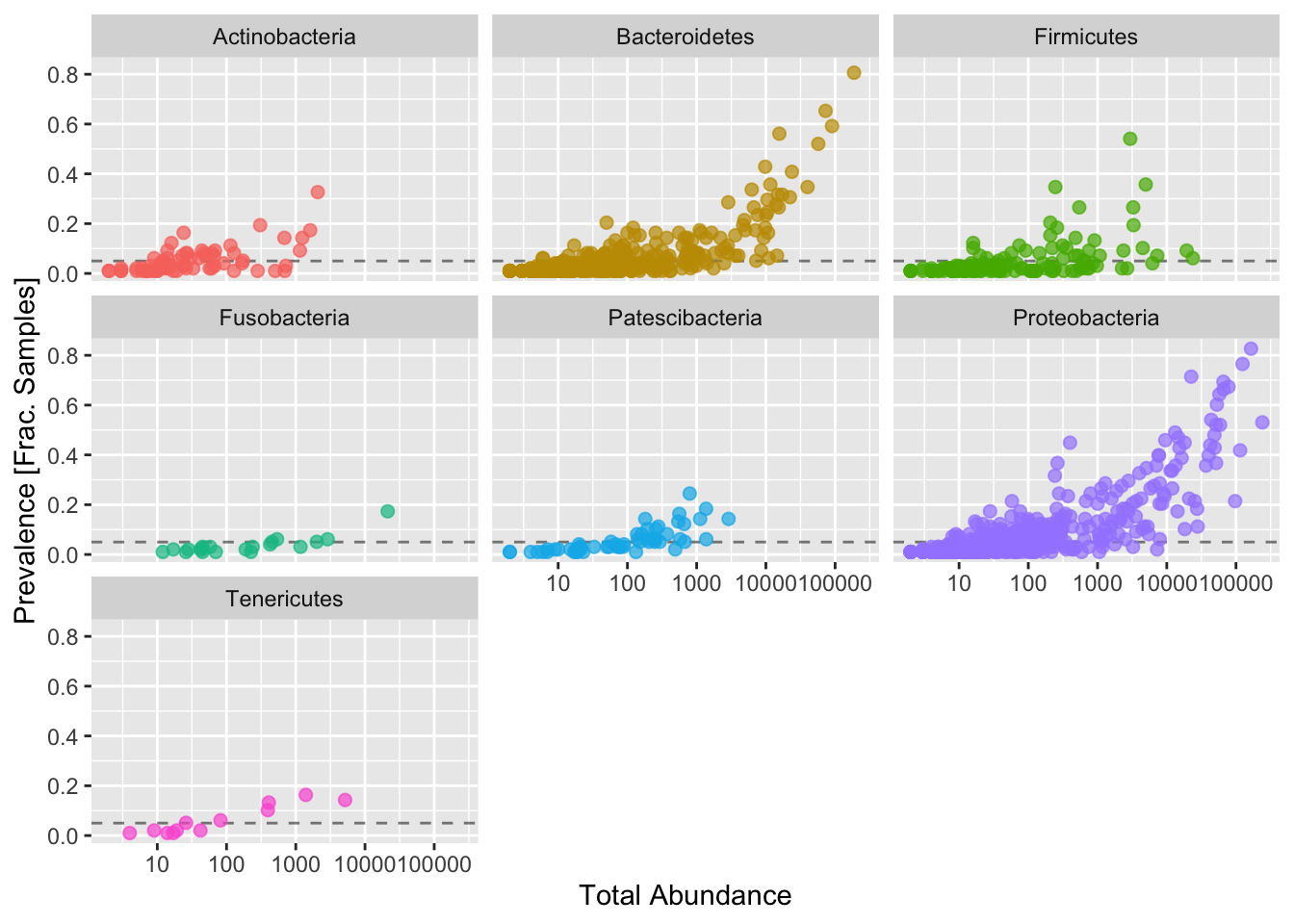
# Definimos el umbral de prevalencia a un 5%
(prevalenceThreshold = 0.05 * nsamples(psd4))## [1] 4.35# Execute prevalence filter, using `prune_taxa()` function
keepTaxa = rownames(prevdf1)[(prevdf1$Prevalence >= prevalenceThreshold)]
(psd5 = prune_taxa(keepTaxa, psd4))## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 136 taxa and 87 samples ]
## sample_data() Sample Data: [ 87 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 136 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 136 tips and 135 internal nodes ]DADA2 usa como nombre de las taxa la secuencia o ASV asociada a un taxon determinado. Esto es conveniente cuando nos interesa la secuencia en nuestros análisis, sin embargo en este práctico solamente vamos a trabajar a nivel de comunidad.
- Por esto, vamos a reemplazar esos nombres con códigos correlativos, lo cual va a hacer las visualizaciones posteriores más entendibles.
# Reemplazamos las secuencias por un nombre genérico
taxa_names(psd5) <- paste0("ASV", seq(ntaxa(psd5)))- Con nuestro objeto phyloseq ya filtrado y listo para usar, podemos gráficar la distribución de read counts por número de muestra de forma de tener una idea sobre la distribución de éstas.
sample_sum_df <- data.frame(sum = sample_sums(psd5))
ggplot(sample_sum_df, aes(x = sum)) +
geom_histogram(color = "black", fill = "grey", binwidth = 2500) +
ggtitle("Distribution of sample sequencing depth") +
xlab("Read counts") +
theme(axis.title.y = element_blank()) 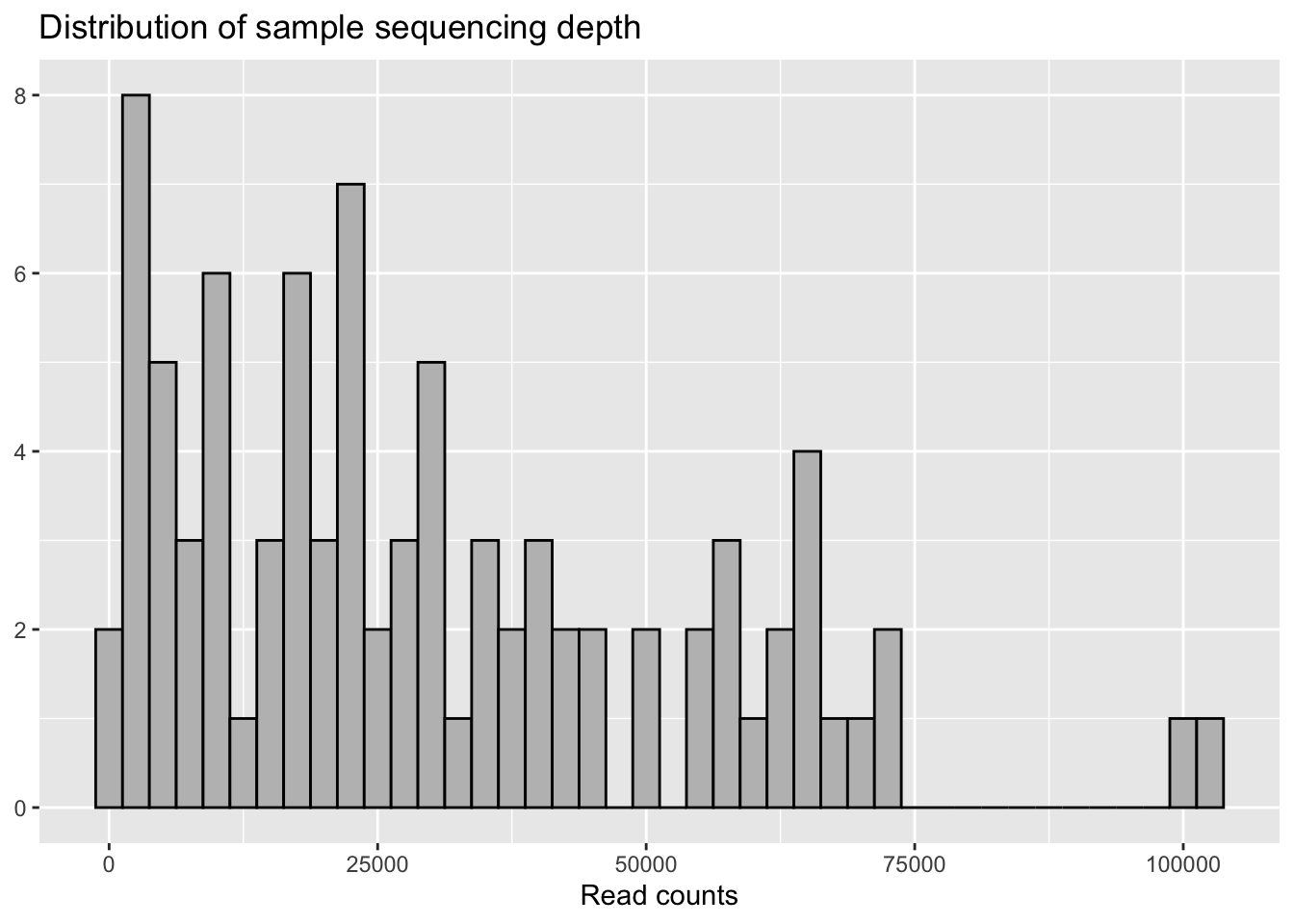
- Finalmente, calculamos curvas de rarefacción para cada muestra, de manera tal que podamos determinar si la profundidad de secuenciación fue sufuciente o si tal vez necesitemos secuenciar más. En otras palabras, este análisis nos permitiría averiguar si al secuenciar más observaríamos más OTUs o ASVs.
# Primero cargamos algunos scripts de manera remota
scripts <- c("graphical_methods.R",
"tree_methods.R",
"plot_merged_trees.R",
"specificity_methods.R",
"ternary_plot.R",
"richness.R",
"edgePCA.R",
"copy_number_correction.R",
"import_frogs.R",
"prevalence.R",
"compute_niche.R")
urls <- paste0("https://raw.githubusercontent.com/mahendra-mariadassou/phyloseq-extended/master/R/", scripts)
for (url in urls) {
source(url)
}# Y graficamos
p <- ggrare(psd5, step = 100, color = "species", label = "sample_ID", se = TRUE)
(p <- p + facet_wrap(~species))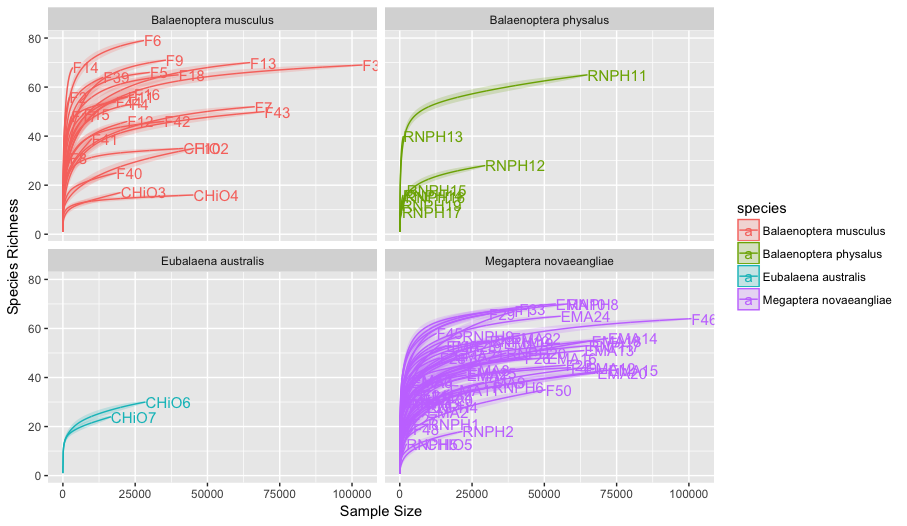
Los gráficos están separados por especies de ballena, azul (Balaenoptera musculus), fin (Balaenoptera physalus), franca (Eubalaena australis), y jorobada (Megaptera novaeangliae) y muestran la cantidad de Taxa (riqueza o diversidad alfa) en función del tamaño muestreal o número de reads. Podemos ver que la mayoría de las muestras de ballena azul y jorobada alcanzan un plateau. Esto significa que el retorno en la inversión disminuye si seguimos secuenciando, o de otra forma, que ya hemos muestreado toda la diversidad de esa muestra. Al contrario, algunas muestras de ballena fin no alcanzaron el plateau, lo cual implica que la diversidad alfa estaría subestimada.
2.2 Estructura y manipulación de un objeto phyloseq
Muchas veces queremos analizar un sub conjunto de las muestras en nuestro objeto phyloseq, o bien, queremos seleccionar ciertos grupos taxonómicos para análisis posteriores. phyloseq nos permite hacer todo tipo de filtros para llevar esto a cabo. Veamos dónde se almacena la información en phyloseq.
- Primero la tabla de cuentas.
| ASV1 | ASV2 | ASV3 | ASV4 | ASV5 | ASV6 | ASV7 | ASV8 | ASV9 | ASV10 | ASV11 | ASV12 | ASV13 | ASV14 | ASV15 | ASV16 | ASV17 | ASV18 | ASV19 | ASV20 | ASV21 | ASV22 | ASV23 | ASV24 | ASV25 | ASV26 | ASV27 | ASV28 | ASV29 | ASV30 | ASV31 | ASV32 | ASV33 | ASV34 | ASV35 | ASV36 | ASV37 | ASV38 | ASV39 | ASV40 | ASV41 | ASV42 | ASV43 | ASV44 | ASV45 | ASV46 | ASV47 | ASV48 | ASV49 | ASV50 | ASV51 | ASV52 | ASV53 | ASV54 | ASV55 | ASV56 | ASV57 | ASV58 | ASV59 | ASV60 | ASV61 | ASV62 | ASV63 | ASV64 | ASV65 | ASV66 | ASV67 | ASV68 | ASV69 | ASV70 | ASV71 | ASV72 | ASV73 | ASV74 | ASV75 | ASV76 | ASV77 | ASV78 | ASV79 | ASV80 | ASV81 | ASV82 | ASV83 | ASV84 | ASV85 | ASV86 | ASV87 | ASV88 | ASV89 | ASV90 | ASV91 | ASV92 | ASV93 | ASV94 | ASV95 | ASV96 | ASV97 | ASV98 | ASV99 | ASV100 | ASV101 | ASV102 | ASV103 | ASV104 | ASV105 | ASV106 | ASV107 | ASV108 | ASV109 | ASV110 | ASV111 | ASV112 | ASV113 | ASV114 | ASV115 | ASV116 | ASV117 | ASV118 | ASV119 | ASV120 | ASV121 | ASV122 | ASV123 | ASV124 | ASV125 | ASV126 | ASV127 | ASV128 | ASV129 | ASV130 | ASV131 | ASV132 | ASV133 | ASV134 | ASV135 | ASV136 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRR6442697 | 0 | 3 | 1 | 4 | 0 | 0 | 5 | 17 | 3 | 0 | 0 | 0 | 1 | 1 | 917 | 1031 | 0 | 3 | 0 | 76 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 15 | 0 | 0 | 534 | 0 | 4 | 0 | 2099 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 158 | 0 | 914 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1014 | 0 | 0 | 322 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 535 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 122 | 0 | 0 | 2 | 0 | 15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 49 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SRR6442698 | 0 | 9 | 537 | 2 | 0 | 0 | 4 | 0 | 8 | 4 | 0 | 0 | 2 | 1 | 2015 | 120 | 0 | 0 | 0 | 36 | 1 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 67 | 0 | 35 | 2 | 306 | 0 | 1 | 0 | 246 | 0 | 0 | 1 | 0 | 0 | 5 | 0 | 0 | 0 | 251 | 0 | 23 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 28 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 3 | 0 | 0 | 178 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 88 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 120 | 0 | 0 | 5 | 0 | 9 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SRR6442699 | 0 | 581 | 0 | 0 | 1 | 0 | 2 | 3 | 6 | 0 | 8 | 0 | 1 | 0 | 2461 | 7 | 0 | 0 | 0 | 1400 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 236 | 0 | 0 | 0 | 575 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 168 | 0 | 1235 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1458 | 0 | 0 | 1208 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 92 | 0 | 0 | 21 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| SRR6442700 | 2 | 8300 | 3828 | 0 | 1 | 0 | 275 | 0 | 9 | 3557 | 8 | 0 | 3324 | 0 | 741 | 1045 | 413 | 6923 | 0 | 253 | 48 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 170 | 0 | 331 | 352 | 0 | 0 | 2913 | 0 | 0 | 2 | 967 | 164 | 0 | 0 | 146 | 361 | 0 | 0 | 0 | 0 | 0 | 2 | 28 | 0 | 17 | 0 | 0 | 0 | 1055 | 1 | 114 | 0 | 0 | 0 | 0 | 0 | 16 | 25 | 0 | 2 | 0 | 79 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 31 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 2 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 15 | 1 | 1 | 4 | 197 | 0 | 0 | 39 | 160 | 0 | 0 | 0 | 0 | 28 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 36 | 0 | 0 | 0 |
| SRR6442701 | 0 | 80 | 526 | 2 | 1 | 88 | 0 | 25 | 249 | 2425 | 182 | 0 | 8 | 0 | 215 | 8 | 7742 | 2455 | 6 | 565 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 18 | 0 | 8 | 65 | 0 | 0 | 0 | 94 | 55 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 9 | 0 | 0 | 0 | 6 | 2550 | 0 | 690 | 1 | 3 | 0 | 0 | 77 | 0 | 85 | 40 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 107 | 2 | 0 | 0 | 0 | 0 | 0 | 72 | 0 | 0 | 0 | 0 | 40 | 101 | 3 | 0 | 108 | 0 | 0 | 0 | 0 | 0 | 241 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| SRR6442702 | 0 | 6 | 49 | 13341 | 3 | 0 | 3583 | 1 | 13608 | 5 | 0 | 0 | 15 | 460 | 3 | 272 | 19137 | 14544 | 0 | 18 | 2 | 0 | 1 | 1 | 3 | 90 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 15 | 0 | 1963 | 1 | 4 | 9 | 6 | 0 | 92 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 9 | 0 | 78 | 0 | 0 | 0 | 766 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 135 | 4 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 66 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 12 | 0 | 31 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
La tabla de cuentas relaciona el nombre de las taxa con las muestras y con el número de reads mapeadas en contra de ellas. Acá el número de reads es directamente proporcional al número de veces que se observa un taxon.
- El otro componente importante es la tabla de taxonomía.
| Kingdom | Phylum | Class | Order | Family | Genus | |
|---|---|---|---|---|---|---|
| ASV1 | Bacteria | Proteobacteria | Gammaproteobacteria | Cardiobacteriales | Cardiobacteriaceae | NA |
| ASV2 | Bacteria | Bacteroidetes | Bacteroidia | Flavobacteriales | Flavobacteriaceae | NA |
| ASV3 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | NA |
| ASV4 | Bacteria | Proteobacteria | Gammaproteobacteria | NA | NA | NA |
| ASV5 | Bacteria | Proteobacteria | Gammaproteobacteria | Xanthomonadales | Xanthomonadaceae | Stenotrophomonas |
| ASV6 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Moraxella |
La tabla de taxonomía relaciona el nombre de las taxa con el linaje taxonómico de éstas, i.e., vincula una variante de secuencia, o ASV, con los rangos taxonómicos desde Reino hasta Género o Especie dependiendo del nivel de resolución del análisis.
- Veamos ahora el otro componente esencial que es la metadata.
| sample_ID | bioproject_accession | study | biosample_accession | experiment | run | SRA_Sample | geo_loc_name | collection_date | sample_type | species | common_name | AvgSpotLen | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRR6442697 | EMA4 | PRJNA428495 | SRP128093 | SAMN08292292 | SRX3533985 | SRR6442697 | SRS2809259 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442698 | EMA3 | PRJNA428495 | SRP128093 | SAMN08292291 | SRX3533984 | SRR6442698 | SRS2809258 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442699 | EMA2 | PRJNA428495 | SRP128093 | SAMN08292284 | SRX3533983 | SRR6442699 | SRS2809257 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442700 | EMA19 | PRJNA428495 | SRP128093 | SAMN08292283 | SRX3533982 | SRR6442700 | SRS2809256 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442701 | EMA21 | PRJNA428495 | SRP128093 | SAMN08292286 | SRX3533981 | SRR6442701 | SRS2809255 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 499 |
| SRR6442702 | EMA20 | PRJNA428495 | SRP128093 | SAMN08292285 | SRX3533980 | SRR6442702 | SRS2809254 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442703 | EMA23 | PRJNA428495 | SRP128093 | SAMN08292288 | SRX3533979 | SRR6442703 | SRS2809252 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442704 | EMA22 | PRJNA428495 | SRP128093 | SAMN08292287 | SRX3533978 | SRR6442704 | SRS2809253 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 499 |
| SRR6442705 | EMA25 | PRJNA428495 | SRP128093 | SAMN08292290 | SRX3533977 | SRR6442705 | SRS2809251 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442706 | EMA24 | PRJNA428495 | SRP128093 | SAMN08292289 | SRX3533976 | SRR6442706 | SRS2809250 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442708 | EMA18 | PRJNA428495 | SRP128093 | SAMN08292282 | SRX3533974 | SRR6442708 | SRS2809249 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442709 | EMA11 | PRJNA428495 | SRP128093 | SAMN08292275 | SRX3533973 | SRR6442709 | SRS2809248 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442710 | EMA12 | PRJNA428495 | SRP128093 | SAMN08292276 | SRX3533972 | SRR6442710 | SRS2809246 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442711 | EMA1 | PRJNA428495 | SRP128093 | SAMN08292273 | SRX3533971 | SRR6442711 | SRS2809245 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442712 | EMA10 | PRJNA428495 | SRP128093 | SAMN08292274 | SRX3533970 | SRR6442712 | SRS2809244 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442713 | EMA15 | PRJNA428495 | SRP128093 | SAMN08292279 | SRX3533969 | SRR6442713 | SRS2809243 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442714 | EMA16 | PRJNA428495 | SRP128093 | SAMN08292280 | SRX3533968 | SRR6442714 | SRS2809242 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442715 | EMA13 | PRJNA428495 | SRP128093 | SAMN08292277 | SRX3533967 | SRR6442715 | SRS2809241 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442716 | EMA14 | PRJNA428495 | SRP128093 | SAMN08292278 | SRX3533966 | SRR6442716 | SRS2809240 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442717 | F1 | PRJNA428495 | SRP128093 | SAMN08292272 | SRX3533965 | SRR6442717 | SRS2809239 | Chile: Chiloe | 2015 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442718 | CHIO5 | PRJNA428495 | SRP128093 | SAMN08292271 | SRX3533964 | SRR6442718 | SRS2809237 | Chile: Chiloe | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442719 | CHiO7 | PRJNA428495 | SRP128093 | SAMN08292270 | SRX3533963 | SRR6442719 | SRS2809238 | Chile: Chiloe | 2017 | skin | Eubalaena australis | southern right whale | 500 |
| SRR6442720 | CHiO6 | PRJNA428495 | SRP128093 | SAMN08292269 | SRX3533962 | SRR6442720 | SRS2809236 | Chile: Chiloe | 2017 | skin | Eubalaena australis | southern right whale | 501 |
| SRR6442721 | CHiO4 | PRJNA428495 | SRP128093 | SAMN08292268 | SRX3533961 | SRR6442721 | SRS2809235 | Chile: Chiloe | 2017 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442722 | CHiO3 | PRJNA428495 | SRP128093 | SAMN08292267 | SRX3533960 | SRR6442722 | SRS2809234 | Chile: Chiloe | 2017 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442723 | CHiO2 | PRJNA428495 | SRP128093 | SAMN08292266 | SRX3533959 | SRR6442723 | SRS2809232 | Chile: Chiloe | 2017 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442725 | F9 | PRJNA428495 | SRP128093 | SAMN08292264 | SRX3533957 | SRR6442725 | SRS2809231 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442726 | F8 | PRJNA428495 | SRP128093 | SAMN08292263 | SRX3533956 | SRR6442726 | SRS2809230 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442727 | F10 | PRJNA428495 | SRP128093 | SAMN08292242 | SRX3533955 | SRR6442727 | SRS2809229 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442731 | RNPH16 | PRJNA428495 | SRP128093 | SAMN08292238 | SRX3533951 | SRR6442731 | SRS2809225 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Balaenoptera physalus | fin whale | 501 |
| SRR6442732 | RNPH15 | PRJNA428495 | SRP128093 | SAMN08292237 | SRX3533950 | SRR6442732 | SRS2809224 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Balaenoptera physalus | fin whale | 501 |
| SRR6442733 | RNPH14 | PRJNA428495 | SRP128093 | SAMN08292236 | SRX3533949 | SRR6442733 | SRS2809223 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Balaenoptera physalus | fin whale | 501 |
| SRR6442734 | RNPH13 | PRJNA428495 | SRP128093 | SAMN08292235 | SRX3533948 | SRR6442734 | SRS2809222 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Balaenoptera physalus | fin whale | 501 |
| SRR6442735 | RNPH12 | PRJNA428495 | SRP128093 | SAMN08292234 | SRX3533947 | SRR6442735 | SRS2809221 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Balaenoptera physalus | fin whale | 501 |
| SRR6442736 | RNPH11 | PRJNA428495 | SRP128093 | SAMN08292233 | SRX3533946 | SRR6442736 | SRS2809220 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Balaenoptera physalus | fin whale | 501 |
| SRR6442737 | F6 | PRJNA428495 | SRP128093 | SAMN08292261 | SRX3533945 | SRR6442737 | SRS2809218 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442738 | F7 | PRJNA428495 | SRP128093 | SAMN08292262 | SRX3533944 | SRR6442738 | SRS2809219 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442739 | F39 | PRJNA428495 | SRP128093 | SAMN08292253 | SRX3533943 | SRR6442739 | SRS2809217 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442740 | F4 | PRJNA428495 | SRP128093 | SAMN08292254 | SRX3533942 | SRR6442740 | SRS2809216 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442741 | F40 | PRJNA428495 | SRP128093 | SAMN08292255 | SRX3533941 | SRR6442741 | SRS2809215 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442742 | F41 | PRJNA428495 | SRP128093 | SAMN08292256 | SRX3533940 | SRR6442742 | SRS2809214 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442743 | F42 | PRJNA428495 | SRP128093 | SAMN08292257 | SRX3533939 | SRR6442743 | SRS2809213 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442744 | F43 | PRJNA428495 | SRP128093 | SAMN08292258 | SRX3533938 | SRR6442744 | SRS2809212 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442745 | F44 | PRJNA428495 | SRP128093 | SAMN08292259 | SRX3533937 | SRR6442745 | SRS2809211 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442746 | F5 | PRJNA428495 | SRP128093 | SAMN08292260 | SRX3533936 | SRR6442746 | SRS2809210 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442747 | RNPH7 | PRJNA428495 | SRP128093 | SAMN08292328 | SRX3533935 | SRR6442747 | SRS2809209 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 499 |
| SRR6442748 | RNPH6 | PRJNA428495 | SRP128093 | SAMN08292327 | SRX3533934 | SRR6442748 | SRS2809208 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442749 | RNPH9 | PRJNA428495 | SRP128093 | SAMN08292330 | SRX3533933 | SRR6442749 | SRS2809207 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442750 | RNPH8 | PRJNA428495 | SRP128093 | SAMN08292329 | SRX3533932 | SRR6442750 | SRS2809206 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442751 | RNPH3 | PRJNA428495 | SRP128093 | SAMN08292324 | SRX3533931 | SRR6442751 | SRS2809205 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442752 | RNPH20 | PRJNA428495 | SRP128093 | SAMN08292323 | SRX3533930 | SRR6442752 | SRS2809204 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442753 | RNPH5 | PRJNA428495 | SRP128093 | SAMN08292326 | SRX3533929 | SRR6442753 | SRS2809203 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442754 | RNPH4 | PRJNA428495 | SRP128093 | SAMN08292325 | SRX3533928 | SRR6442754 | SRS2809202 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 499 |
| SRR6442755 | F16 | PRJNA428495 | SRP128093 | SAMN08292248 | SRX3533927 | SRR6442755 | SRS2809201 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442756 | F15 | PRJNA428495 | SRP128093 | SAMN08292247 | SRX3533926 | SRR6442756 | SRS2809199 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442757 | F18 | PRJNA428495 | SRP128093 | SAMN08292250 | SRX3533925 | SRR6442757 | SRS2809200 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442758 | F17 | PRJNA428495 | SRP128093 | SAMN08292249 | SRX3533924 | SRR6442758 | SRS2809198 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442759 | F12 | PRJNA428495 | SRP128093 | SAMN08292244 | SRX3533923 | SRR6442759 | SRS2809197 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442760 | F11 | PRJNA428495 | SRP128093 | SAMN08292243 | SRX3533922 | SRR6442760 | SRS2809196 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442761 | F14 | PRJNA428495 | SRP128093 | SAMN08292246 | SRX3533921 | SRR6442761 | SRS2809195 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442762 | F13 | PRJNA428495 | SRP128093 | SAMN08292245 | SRX3533920 | SRR6442762 | SRS2809193 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 501 |
| SRR6442763 | F3 | PRJNA428495 | SRP128093 | SAMN08292252 | SRX3533919 | SRR6442763 | SRS2809194 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442764 | F2 | PRJNA428495 | SRP128093 | SAMN08292251 | SRX3533918 | SRR6442764 | SRS2809192 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 |
| SRR6442765 | RNPH10 | PRJNA428495 | SRP128093 | SAMN08292321 | SRX3533917 | SRR6442765 | SRS2809191 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442766 | RNPH2 | PRJNA428495 | SRP128093 | SAMN08292322 | SRX3533916 | SRR6442766 | SRS2809190 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442767 | F50 | PRJNA428495 | SRP128093 | SAMN08292319 | SRX3533915 | SRR6442767 | SRS2809189 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 499 |
| SRR6442768 | RNPH1 | PRJNA428495 | SRP128093 | SAMN08292320 | SRX3533914 | SRR6442768 | SRS2809188 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442769 | F48 | PRJNA428495 | SRP128093 | SAMN08292317 | SRX3533913 | SRR6442769 | SRS2809187 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442770 | F49 | PRJNA428495 | SRP128093 | SAMN08292318 | SRX3533912 | SRR6442770 | SRS2809186 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442771 | F46 | PRJNA428495 | SRP128093 | SAMN08292315 | SRX3533911 | SRR6442771 | SRS2809185 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442774 | F45 | PRJNA428495 | SRP128093 | SAMN08292314 | SRX3533908 | SRR6442774 | SRS2809181 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442775 | F27 | PRJNA428495 | SRP128093 | SAMN08292306 | SRX3533907 | SRR6442775 | SRS2809180 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442776 | F26 | PRJNA428495 | SRP128093 | SAMN08292305 | SRX3533906 | SRR6442776 | SRS2809179 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442779 | F31 | PRJNA428495 | SRP128093 | SAMN08292310 | SRX3533903 | SRR6442779 | SRS2809176 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 499 |
| SRR6442780 | F30 | PRJNA428495 | SRP128093 | SAMN08292309 | SRX3533902 | SRR6442780 | SRS2809175 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442781 | F29 | PRJNA428495 | SRP128093 | SAMN08292308 | SRX3533901 | SRR6442781 | SRS2809174 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442782 | F28 | PRJNA428495 | SRP128093 | SAMN08292307 | SRX3533900 | SRR6442782 | SRS2809183 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442783 | F33 | PRJNA428495 | SRP128093 | SAMN08292312 | SRX3533899 | SRR6442783 | SRS2809173 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442784 | F32 | PRJNA428495 | SRP128093 | SAMN08292311 | SRX3533898 | SRR6442784 | SRS2809172 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442785 | F22 | PRJNA428495 | SRP128093 | SAMN08292301 | SRX3533897 | SRR6442785 | SRS2809171 | Chile: Estrecho_Magallanes | 2010 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442786 | F23 | PRJNA428495 | SRP128093 | SAMN08292302 | SRX3533896 | SRR6442786 | SRS2809170 | Chile: Estrecho_Magallanes | 2010 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442787 | EMA9 | PRJNA428495 | SRP128093 | SAMN08292297 | SRX3533895 | SRR6442787 | SRS2809169 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442788 | F19 | PRJNA428495 | SRP128093 | SAMN08292298 | SRX3533894 | SRR6442788 | SRS2809168 | Chile: Estrecho_Magallanes | 2010 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442789 | F20 | PRJNA428495 | SRP128093 | SAMN08292299 | SRX3533893 | SRR6442789 | SRS2809167 | Chile: Estrecho_Magallanes | 2010 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442790 | F21 | PRJNA428495 | SRP128093 | SAMN08292300 | SRX3533892 | SRR6442790 | SRS2809166 | Chile: Estrecho_Magallanes | 2010 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442792 | EMA6 | PRJNA428495 | SRP128093 | SAMN08292294 | SRX3533890 | SRR6442792 | SRS2809164 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442794 | EMA8 | PRJNA428495 | SRP128093 | SAMN08292296 | SRX3533888 | SRR6442794 | SRS2809162 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
Finalmente, tenemos el árbol filogenético, que es opcional en phyloseq, que nos muestra las relaciones evolutivas entre las taxa de todas las muestras. Es opcional porque normalmente cuando hacemos shotgun metagenomics no contamos con un marcador universal y por lo tanto no hay filogenia.
- Podemos graficar simplemente la filogenia con la función
plot_tree.
# Esta es la filogenia asociada a las taxa en nuestro objeto phyloseq
plot_tree(psd5, method = "treeonly", ladderize = "left")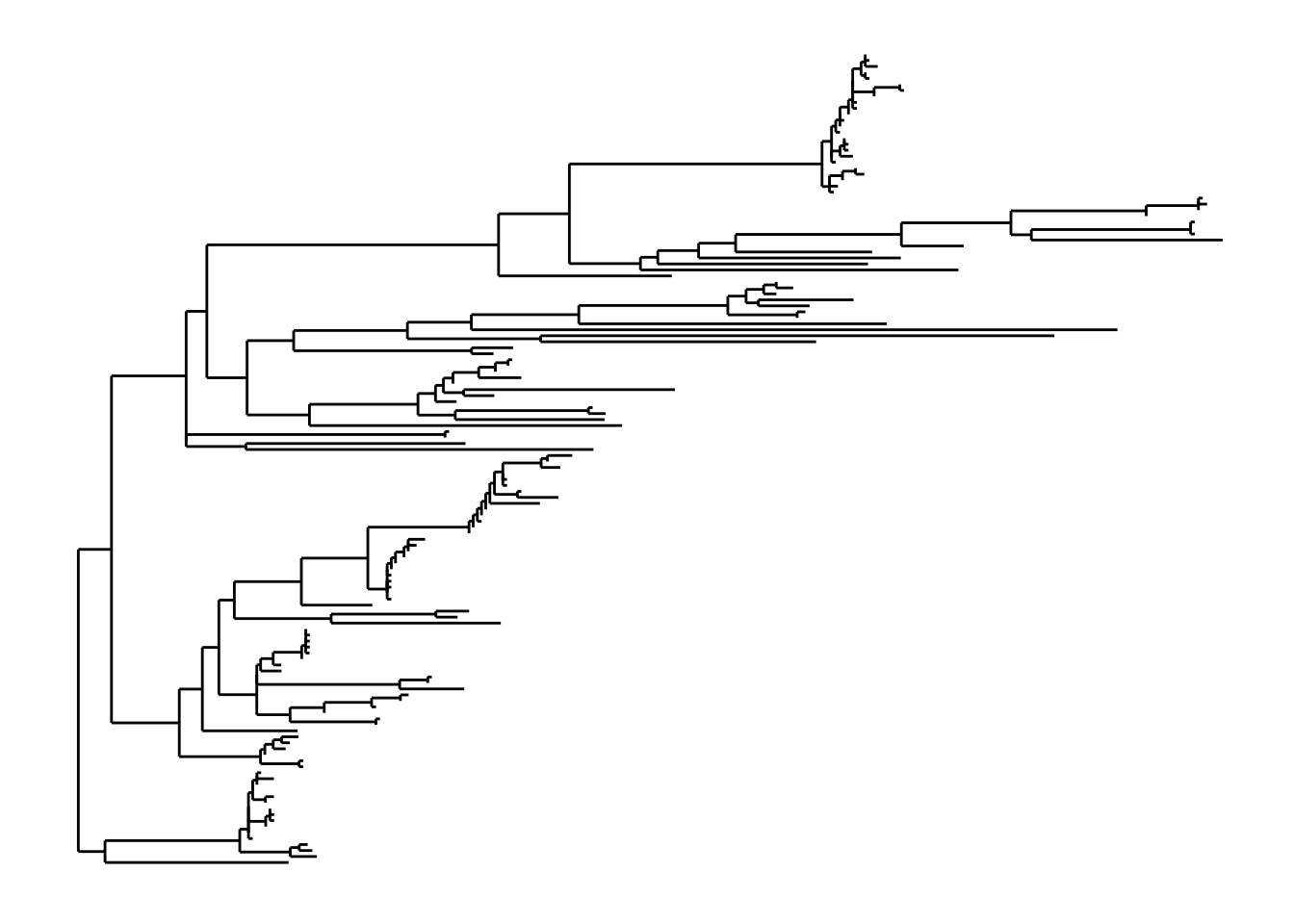
Ahora, el objeto phyloseq se ha vuelto una suerte de estándar en la industria ya que otros paquetes ahora usan esta estructura de datos para sus propias funciones. Uno de esos paquetes es microbiome y ampvis. Podemos fácilmente obtener un resumen global de nuestro objeto phyloseq usando la función summarize_phyloseq.
- Primero cargamos el paquete con
library(microbiome).
summarize_phyloseq(psd5)## Compositional = NO
## 1] Min. number of reads = 1123
## 2] Max. number of reads = 103541
## 3] Total number of reads = 2606004
## 4] Average number of reads = 29954.0689655172
## 5] Median number of reads = 23576
## 7] Sparsity = 0.679428668018932
## 6] Any OTU sum to 1 or less? NO
## 8] Number of singletons = 0
## 9] Percent of OTUs that are singletons 0
## 10] Number of sample variables are: 13
## sample_ID
## bioproject_accession
## study
## biosample_accession
## experiment
## run
## SRA_Sample
## geo_loc_name
## collection_date
## sample_type
## species
## common_name
## AvgSpotLenEste comando nos muestra el mínimo y máximo de reads, número total y promedio de reads, etc. También muestra los encabezados de las columans en la tabla de metadata.
- Veamos ahora una tabla que mezcle metadata, taxonomíaa y abundancia del taxon más abundante de cada muestra.
df <- psmelt(psd5)| OTU | Sample | Abundance | sample_ID | bioproject_accession | study | biosample_accession | experiment | run | SRA_Sample | geo_loc_name | collection_date | sample_type | species | common_name | AvgSpotLen | Kingdom | Phylum | Class | Order | Family | Genus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | ASV1 | SRR6442744 | 65451 | F43 | PRJNA428495 | SRP128093 | SAMN08292258 | SRX3533938 | SRR6442744 | SRS2809212 | Chile: Chiloe | 2016 | skin | Balaenoptera musculus | blue whale | 501 | Bacteria | Proteobacteria | Gammaproteobacteria | Cardiobacteriales | Cardiobacteriaceae | NA |
| 8009 | ASV6 | SRR6442747 | 57107 | RNPH7 | PRJNA428495 | SRP128093 | SAMN08292328 | SRX3533935 | SRR6442747 | SRS2809209 | Chile: Reserva_Nacional_Pinguino_de_Humboldt | 2017 | skin | Megaptera novaeangliae | humpback whale | 499 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Moraxella |
| 39 | ASV1 | SRR6442738 | 46464 | F7 | PRJNA428495 | SRP128093 | SAMN08292262 | SRX3533944 | SRR6442738 | SRS2809219 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 501 | Bacteria | Proteobacteria | Gammaproteobacteria | Cardiobacteriales | Cardiobacteriaceae | NA |
| 3702 | ASV14 | SRR6442771 | 44378 | F46 | PRJNA428495 | SRP128093 | SAMN08292315 | SRX3533911 | SRR6442771 | SRS2809185 | Chile: Estrecho_Magallanes | 2016 | skin | Megaptera novaeangliae | humpback whale | 501 | Bacteria | Bacteroidetes | Bacteroidia | Flavobacteriales | Flavobacteriaceae | Tenacibaculum |
| 6139 | ASV4 | SRR6442763 | 42681 | F3 | PRJNA428495 | SRP128093 | SAMN08292252 | SRX3533919 | SRR6442763 | SRS2809194 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 | Bacteria | Proteobacteria | Gammaproteobacteria | NA | NA | NA |
| 4144 | ASV19 | SRR6442763 | 37149 | F3 | PRJNA428495 | SRP128093 | SAMN08292252 | SRX3533919 | SRR6442763 | SRS2809194 | Chile: Chiloe | 2015 | skin | Balaenoptera musculus | blue whale | 500 | Bacteria | Proteobacteria | Gammaproteobacteria | Cardiobacteriales | Cardiobacteriaceae | NA |
- También es importante tener una visión de cómo se distribuyen las muestras de acuerdo a la metadata. En este ejemplo, graficamos la frecuencia de muestras de acuerdo a la ubicación geográfica (geo_loc_name) y a la especie de ballena de donde la muestra fue obtenida (species).
res <- plot_frequencies(sample_data(psd5), "geo_loc_name", "species")
print(res$plot)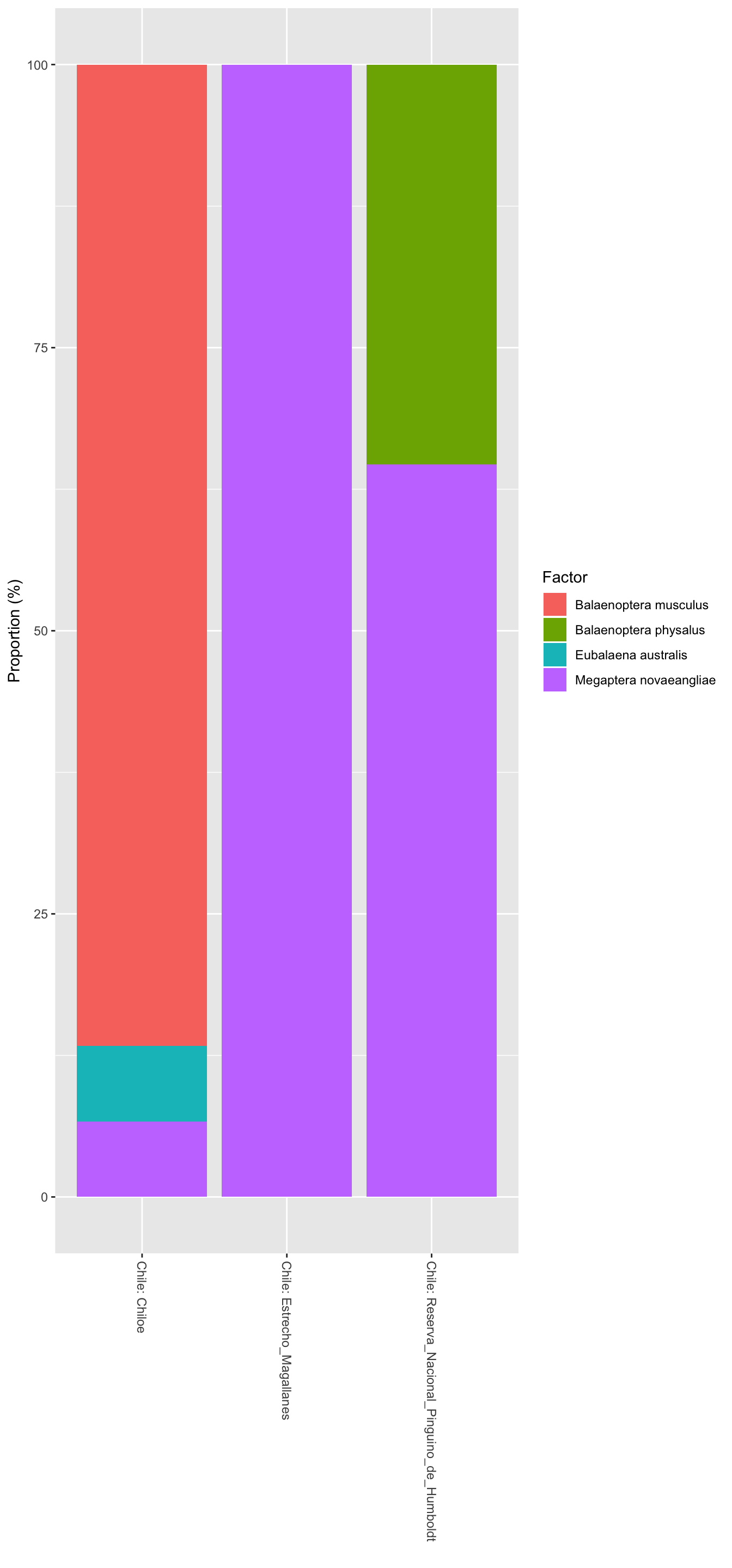
| Groups | Factor | n | pct |
|---|---|---|---|
| Chile: Chiloe | Balaenoptera musculus | 26 | 86.67 |
| Chile: Chiloe | Eubalaena australis | 2 | 6.67 |
| Chile: Chiloe | Megaptera novaeangliae | 2 | 6.67 |
| Chile: Estrecho_Magallanes | Megaptera novaeangliae | 40 | 100.00 |
| Chile: Reserva_Nacional_Pinguino_de_Humboldt | Balaenoptera physalus | 6 | 35.29 |
| Chile: Reserva_Nacional_Pinguino_de_Humboldt | Megaptera novaeangliae | 11 | 64.71 |
Ahora veamos cómo podemos filtrar y hacer subsetting de un objeto phyloseq. Esto lo hacemos con tres grupos de funciones, i.e., filter, subset, y prune. Filtrar se refiere a filtrar según alguna regla lógica. Ya lo hicimos en la parte de control de calidad cuando llamamos la función filter_taxa(psd1, function(x) mean(x) > 1e-5, TRUE). Acá le pedíamos a la función filter_taxa que sobre el objeto psd5, calculara la media de read counts para cada taxa y si este resultado era menor que 1e-5, lo eliminara. Veamos un ejemplo diferente y filtremos según abundancia.
- Primero transformamos en abundancia relativa y luego filtramos.
# Transformamos las cuentas en porcentaje
psd5r = transform_sample_counts(psd5, function(x) x / sum(x) )
# Filtramos las taxa con una abundancia inferior al 1%
(psd5r.filtrado = filter_taxa(psd5r, function(x) sum(x) > 1, TRUE))## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 24 taxa and 87 samples ]
## sample_data() Sample Data: [ 87 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 24 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 24 tips and 23 internal nodes ]¿Cuántas taxa permanecen en nuestro objeto phyloseq? Con una operación tan simple como la que acabamos de aplicar, nos damos cuenta que la mayoría de las taxa presentes en nuestras muestras están en muy baja abundancia.
- Ahora imaginemos la situación donde queremos filtrar nuestro objeto pero en función de un taxon en específico.
| Genus | |
|---|---|
| ASV1 | NA |
| ASV2 | NA |
| ASV3 | NA |
| ASV4 | NA |
| ASV5 | Stenotrophomonas |
| ASV6 | Moraxella |
| ASV7 | Tenacibaculum |
| ASV8 | Klebsiella |
| ASV9 | Tenacibaculum |
| ASV10 | NA |
| ASV11 | NA |
| ASV12 | Pseudomonas |
| ASV13 | NA |
| ASV15 | NA |
| ASV16 | Moraxella |
| ASV17 | Moraxella |
| ASV18 | NA |
| ASV19 | NA |
| ASV20 | NA |
| ASV22 | Pseudomonas |
| ASV23 | Tenacibaculum |
| ASV24 | Achromobacter |
| ASV25 | Catenococcus |
| ASV28 | Escherichia/Shigella |
# Ahora filtramos de acuerdo a _Moraxella_
(subset_taxa(psd5r.filtrado, Genus=="Moraxella") -> psd5r.filtrado.moraxella)## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 3 taxa and 87 samples ]
## sample_data() Sample Data: [ 87 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 3 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 3 tips and 2 internal nodes ]# También podríamos todo lo que NO es _Moraxella_
(subset_taxa(psd5r.filtrado, Genus!="Moraxella") -> psd5r.filtrado.NoMoraxella)## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 10 taxa and 87 samples ]
## sample_data() Sample Data: [ 87 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 10 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 10 tips and 9 internal nodes ]- Otra manera de filtrar un objeto
phyloseqes en base a algún atributo presente ensample_data. Por ejemplo, con estos datos uno podría querer estudiar el microbioma de las ballenas por separado. Para esto crearíamos tres objetosphyloseqa partir depsd5.
(psd5.blue = subset_samples(psd5, species == "Balaenoptera musculus"))## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 136 taxa and 26 samples ]
## sample_data() Sample Data: [ 26 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 136 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 136 tips and 135 internal nodes ](psd5.fin = subset_samples(psd5, species == "Balaenoptera physalus"))## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 136 taxa and 6 samples ]
## sample_data() Sample Data: [ 6 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 136 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 136 tips and 135 internal nodes ](psd5.joro = subset_samples(psd5, species == "Megaptera novaeangliae"))## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 136 taxa and 53 samples ]
## sample_data() Sample Data: [ 53 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 136 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 136 tips and 135 internal nodes ]- Alternativamente, podríamos decicir estudiar solo tres de las cuatro especies de ballenas que tenemos representadas en
psd5.
psd5 = subset_samples(subset_samples(psd5, species != "Eubalaena australis"))- El comando
prune_samples()también es muy usado ya que nos permite usar un vector con las muestras que queremos mantener (similar asubset_samples) o un vector lógico donde las muestras que queremos mantener son verdaderas.
# Primero seleccionamos solo el género _Moraxella_
subset_taxa(psd5, Genus=="Moraxella") -> psd5.moraxella
# Luego nos quedamos con las muestras que solo cumplen con la condición, i,e, que poseen una abundancia de _Moraxella_ de más de 5 reads
prune_samples(sample_sums(psd5.moraxella)>=5, psd5.moraxella) -> psd5.moraxella
# Y finalmente visualizamos los resultados mapeados en el árbol filogenético
plot_tree(psd5.moraxella, color="species", shape="Family", label.tips="Genus", size="abundance")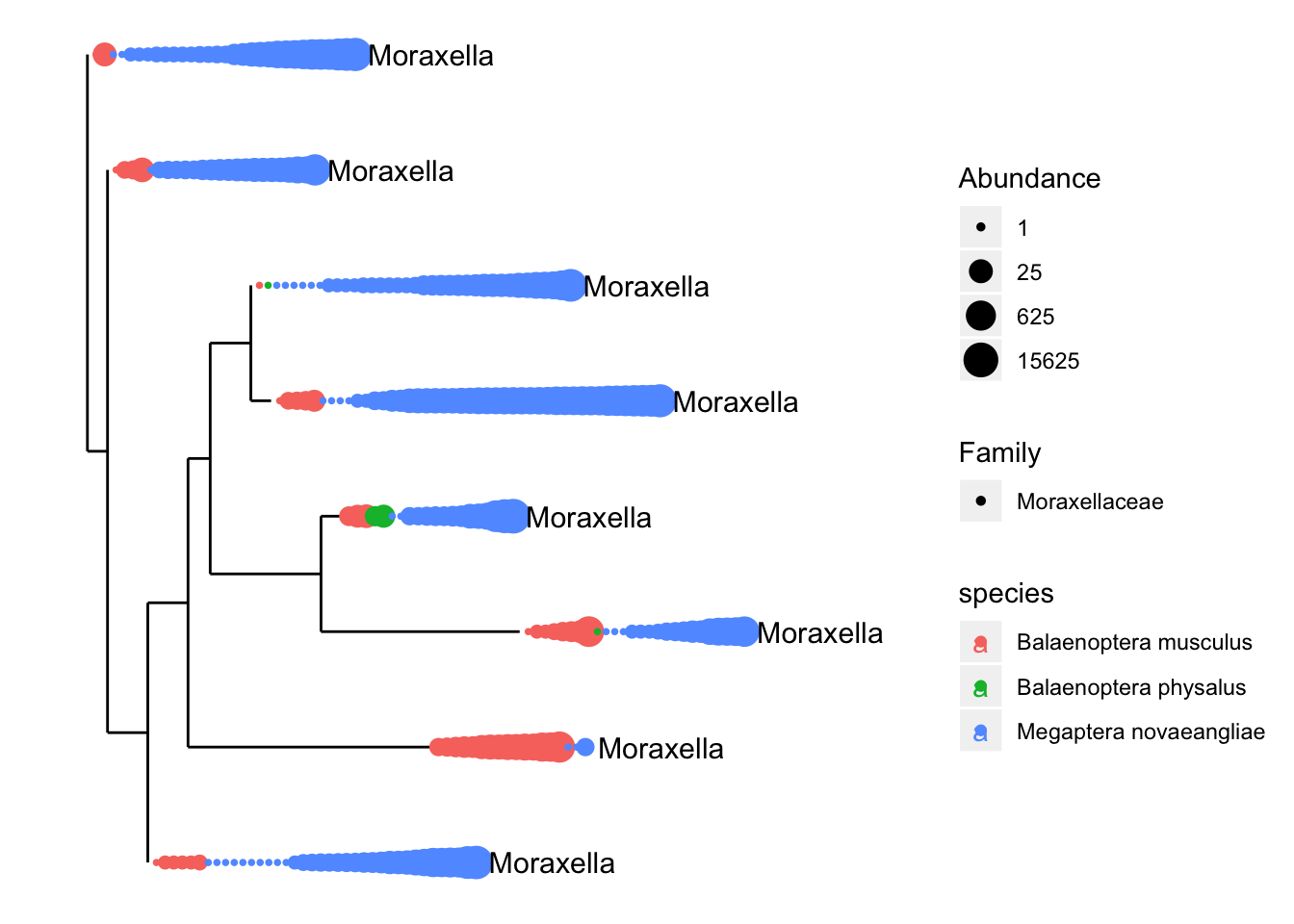
Inmediatamente podemos apreciar que la distribución de Moraxella es mayor en ballena jorobada que en las otras dos especies, azul y fin.
- Otra situación muy común ocurre cuando queremos remover contaminantes u otras taxa no deseadas. Esto se puede hacer fácilmente con el comando
prune_taxa.
# Primero definimos las taxa que no queremos
badTaxa = c("ASV134", "ASV104", "ASV68")
# Creamos una lista con todos los nombres de los taxa presentes en el objeto `psd5`
allTaxa = taxa_names(psd5)
# Nos quedamos con la diferencia entre badTaxa y allTaxa
keepTaxa <- allTaxa[!(allTaxa %in% badTaxa)]
#Ejecutamos `prune_taxa` sobre psd5
(psd5.prune = prune_taxa(keepTaxa, psd5))## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 133 taxa and 85 samples ]
## sample_data() Sample Data: [ 85 samples by 13 sample variables ]
## tax_table() Taxonomy Table: [ 133 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 133 tips and 132 internal nodes ]Para finalizar esta sección, un par de funciones muy útiles en phyloseq son tax_glom() y tip_glom(). Ambas funciones tratan de agrupar o aglomerar un objeto de acuerdo a alguna propiedad, de esta manera simplificándolo. Por ejemplo, es muy probable que uno tenga varias ASVs del mismo género ya que si bien a nivel de secuencia son diferentes, estas corresponden al mismo género. En cierta forma ya lo vimos cuando seleccionamos el género Moraxella. El objeto resultante tenía ocho taxa, todas ellas Moraxella.
- Para hacer visualizaciones y otros análisis puede ser conveniente colapsar o aglomerar estas secuencias del mismo género u otro rango taxonómico. Al mismo tiempo,
tip_glomrealiza una función similar pero basándose en una “altura” arbitraria en el árbol filogenético.
# Primero aglomeramos por género
psd5.genus = tax_glom(psd5, "Genus", NArm = FALSE)
# Luego por altura en el árbol filogenético
h1 = 0.4
psd5.tip = tip_glom(psd5, h = h1)
# Grafiquemos una comparación para visualizar las diferencias
multiPlotTitleTextSize = 15
p2tree = plot_tree(psd5, method = "treeonly",
ladderize = "left",
title = "Sin aglomeración") +
theme(plot.title = element_text(size = multiPlotTitleTextSize))
p3tree = plot_tree(psd5.genus, method = "treeonly",
ladderize = "left", title = "A nivel de género") +
theme(plot.title = element_text(size = multiPlotTitleTextSize))
p4tree = plot_tree(psd5.tip, method = "treeonly",
ladderize = "left", title = "Por altura") +
theme(plot.title = element_text(size = multiPlotTitleTextSize))
# Graficamos los árboles juntos
grid.arrange(nrow = 1, p2tree, p3tree, p4tree)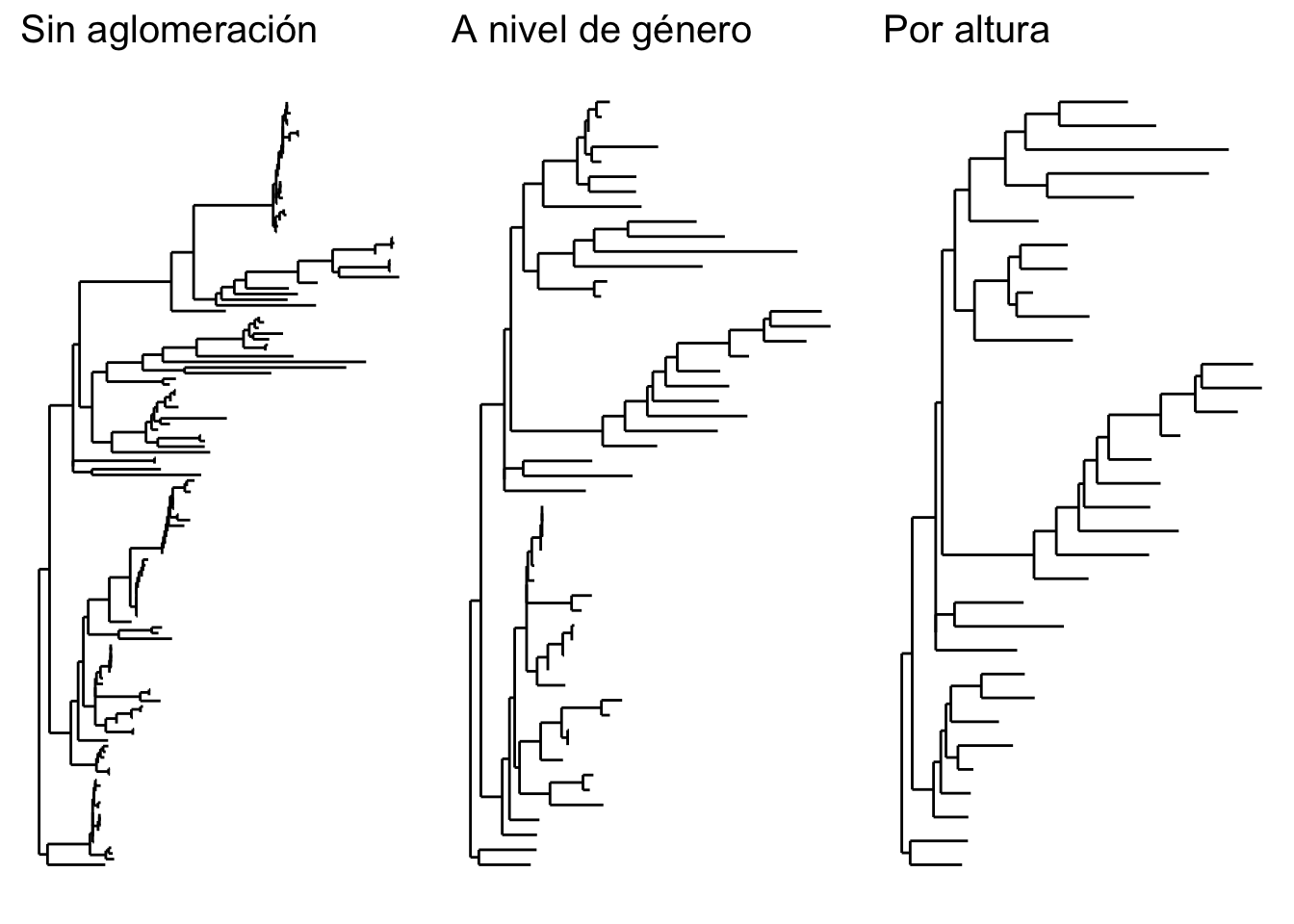
3 Introducción al análisis de diversidad
¿Qué entendemos por diversidad? Al menos podemos conceptualizar diversidad a dos niveles: diversidad genética o morfológica, y biodiversidad. En el estudio de comunidades, tomamos prestado el concepto de biodiversidad de ecología de comunidades donde estamos interesados en la riqueza de especies (número de especies diferentes en una comunidad o diversidad alfa), en las diferencias y similitudes entre comunidades (diversidad beta), y en algunos casos en la diversidad total de un región o paisaje ecológico (landscape; diversidad gama).

En la figura observamos la diversidad de aves en dos regiones, X e Y, y cuatro sitios, 1-4 (figura tomada de Community Ecology de Mittelbach). La diversidad alfa es mayor en los sitios 1 y 3 con 5 especies cada uno. La diversidad beta mide la cantidad de cambio o turnover de especies entre sitios. En la figura, la región Y tiene una diversidad beta mayor que la región X porque el cambio o turnover de especies entre el sitio 2 y el 4 es mayor que entre el sitio 1 y el 3 (que tienen las mismas 5 especies). La diversidad gama mide la diversidad total dentro de una región, por lo tanto en nuestro ejemplo la diversidad gama es mayor en la región Y porque contiene 6 especies en total versus la región X que tiene 5 especies.
3.1 Medidas de riqueza, uniformidad, dominancia, diversidad filogenética (diversidad alfa)
En el contexto metagenómico, medimos diversidad alfa usando una serie de medidas prestadas de ecología que nos permiten caracterizar una comunidad microbiana. phyloseq tiene una función muy útil que nos permite calcular y graficar hasta siete medidas, i .e., Observed (simplemente el número de taxa o riqueza), Chao1 (la riqueza ajustada por probabilidad de no observar especies), ACE (riqueza que toma en cuenta la abundancia relativa), Shannon (abundancia relativa de taxa), Simpson (1 - la probabilidad de que observemos aleatoriamente dos bacterias en una comunidad y que pertenezcan a diferentes especies ), Inverse Simpson ( 1 / Simpson), y Fisher (riqueza tomando en cuenta abundancia).
- En
phyloseqsimplemente llamamos la funciónplot_richnessy podemos visualizar las medidas de diversidad.
plot_richness(psd5, color = "species", x = "species", measures = c("Observed", "Chao1", "Shannon")) + geom_boxplot(aes(fill = species), alpha=.7) + scale_color_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f")) + scale_fill_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f"))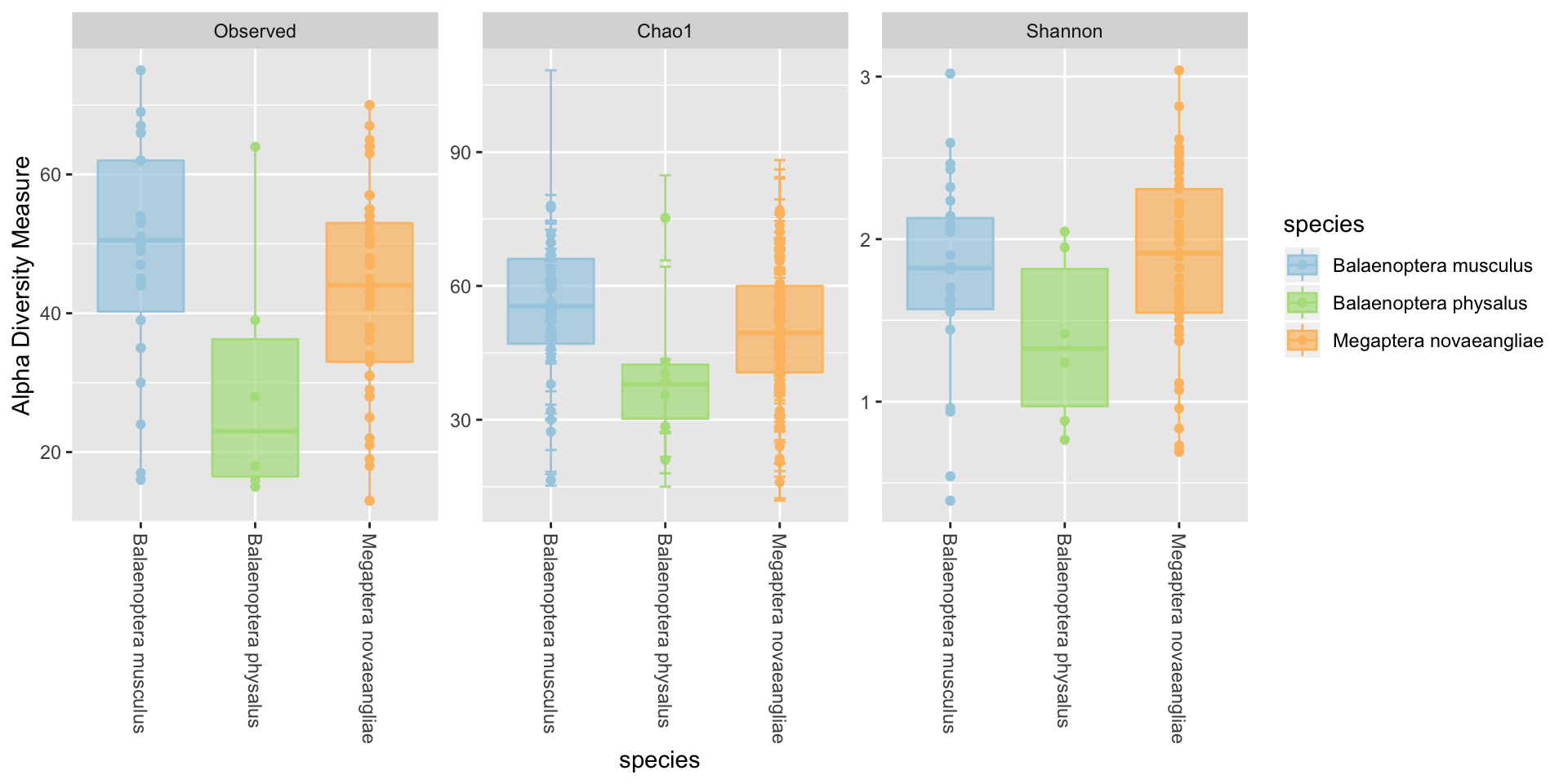
En el ejemplo, solo graficamos Observed, Chao1 y Shannon usando el argumento measures = c("Observed", "Chao1", "Shannon"). Si quisieramos obtener todas las medidas simplemente eliminamos este argumento y por defecto phyloseq graficará todo.
¿Hay un efecto significativo de la diversidad alfa según especie de ballena? Eso lo podríamos probar rápidamente con un análisis de varianza (ANOVA). Para este ejemplo utilicemos otra medida de diversidad, una que phyloseq no incorpora. Faith’s Phylogenetic Diversity es un índice introducido por Daniel Faith en 1992 que no solo considera número de especies sino que también considera qué tanto se parecen estas especies filogenéticamente. Esto es muy relevante porque nos entrega una medida rápida para evaluar prioridades de conservación de ecosistemas, o si se trata de comunidades microbianas, donde tenemos mayor probabilidad de encontrar funciones génicas novedosas.
# Guardamos un dataframe con las medidas de diversidad alfa
alpha_pd <- estimate_pd(psd5)
# Combinamos la metadata con alpha.diversity
data <- cbind(sample_data(psd5), alpha_pd)
# Y calculamos un ANOVA
psd5.anova <- aov(PD ~ species, data)
# install.packages("xtable")
library(xtable)
psd5.anova.table <- xtable(psd5.anova)| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| species | 2 | 40.46002 | 20.23001 | 5.21047 | 0.00741 |
| Residuals | 82 | 318.37078 | 3.88257 | NA | NA |
- El paquete
microbiomeofrece otras herramientas para evaluar diversidad que son accesibles fácilmente a través de su funciónglobal.
tab <- global(psd5, index = "all")
head(tab)## richness_0 richness_20 richness_50 richness_80 observed
## SRR6442697 31 31 31 16 31
## SRR6442698 38 38 38 17 38
## SRR6442699 25 25 25 12 25
## SRR6442700 53 53 53 36 53
## SRR6442701 48 48 48 32 48
## SRR6442702 42 42 42 22 42
## diversities_inverse_simpson diversities_gini_simpson
## SRR6442697 6.948971 0.8560938
## SRR6442698 3.673899 0.7278096
## SRR6442699 6.461545 0.8452383
## SRR6442700 7.707637 0.8702586
## SRR6442701 4.520999 0.7788099
## SRR6442702 4.873575 0.7948118
## diversities_shannon diversities_fisher diversities_coverage
## SRR6442697 2.181249 4.100949 3
## SRR6442698 1.913672 5.781418 2
## SRR6442699 2.043740 3.117388 3
## SRR6442700 2.412447 6.104153 3
## SRR6442701 2.038536 5.949163 2
## SRR6442702 1.759212 4.346443 3
## evenness_camargo evenness_pielou evenness_simpson evenness_evar
## SRR6442697 0.04991266 0.6351942 0.05109538 0.09528851
## SRR6442698 0.04179604 0.5260830 0.02701396 0.13983565
## SRR6442699 0.04419760 0.6349235 0.04751136 0.07442449
## SRR6442700 0.06210963 0.6076245 0.05667380 0.08087544
## SRR6442701 0.04676204 0.5265900 0.03324264 0.10368220
## SRR6442702 0.03423354 0.4706709 0.03583511 0.07058635
## evenness_bulla dominance_dbp dominance_dmn dominance_absolute
## SRR6442697 0.09213899 0.2669465 0.3980669 2099
## SRR6442698 0.11343557 0.4878935 0.6179177 2015
## SRR6442699 0.07294103 0.2597910 0.4137021 2461
## SRR6442700 0.14903074 0.2305235 0.4228024 8300
## SRR6442701 0.13473186 0.4078386 0.5421693 7742
## SRR6442702 0.06516802 0.2799936 0.4927869 19137
## dominance_relative dominance_simpson dominance_core_abundance
## SRR6442697 0.2669465 0.1439062 0.2618593
## SRR6442698 0.4878935 0.2721904 0.6714286
## SRR6442699 0.2597910 0.1547617 0.4718674
## SRR6442700 0.2305235 0.1297414 0.6071379
## SRR6442701 0.4078386 0.2211901 0.2298899
## SRR6442702 0.2799936 0.2051882 0.4588576
## dominance_gini rarity_log_modulo_skewness rarity_low_abundance
## SRR6442697 0.9500873 2.059940 0.009538344
## SRR6442698 0.9582040 2.060444 0.015012107
## SRR6442699 0.9558024 2.038428 0.004011401
## SRR6442700 0.9378904 2.051077 0.009359811
## SRR6442701 0.9532380 2.054745 0.006900911
## SRR6442702 0.9657665 2.053781 0.009861298
## rarity_noncore_abundance rarity_rare_abundance
## SRR6442697 0.131756327 0.131756327
## SRR6442698 0.095883777 0.095883777
## SRR6442699 0.137337697 0.137337697
## SRR6442700 0.012887099 0.012887099
## SRR6442701 0.031607228 0.031607228
## SRR6442702 0.003131035 0.003131035| richness_0 | richness_20 | richness_50 | richness_80 | observed | diversities_inverse_simpson | diversities_gini_simpson | diversities_shannon | diversities_fisher | diversities_coverage | evenness_camargo | evenness_pielou | evenness_simpson | evenness_evar | evenness_bulla | dominance_dbp | dominance_dmn | dominance_absolute | dominance_relative | dominance_simpson | dominance_core_abundance | dominance_gini | rarity_log_modulo_skewness | rarity_low_abundance | rarity_noncore_abundance | rarity_rare_abundance | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRR6442697 | 31 | 31 | 31 | 16 | 31 | 6.95 | 0.86 | 2.18 | 4.10 | 3 | 0.05 | 0.64 | 0.05 | 0.10 | 0.09 | 0.27 | 0.40 | 2099 | 0.27 | 0.14 | 0.26 | 0.95 | 2.06 | 0.01 | 0.13 | 0.13 |
| SRR6442698 | 38 | 38 | 38 | 17 | 38 | 3.67 | 0.73 | 1.91 | 5.78 | 2 | 0.04 | 0.53 | 0.03 | 0.14 | 0.11 | 0.49 | 0.62 | 2015 | 0.49 | 0.27 | 0.67 | 0.96 | 2.06 | 0.02 | 0.10 | 0.10 |
| SRR6442699 | 25 | 25 | 25 | 12 | 25 | 6.46 | 0.85 | 2.04 | 3.12 | 3 | 0.04 | 0.63 | 0.05 | 0.07 | 0.07 | 0.26 | 0.41 | 2461 | 0.26 | 0.15 | 0.47 | 0.96 | 2.04 | 0.00 | 0.14 | 0.14 |
| SRR6442700 | 53 | 53 | 53 | 36 | 53 | 7.71 | 0.87 | 2.41 | 6.10 | 3 | 0.06 | 0.61 | 0.06 | 0.08 | 0.15 | 0.23 | 0.42 | 8300 | 0.23 | 0.13 | 0.61 | 0.94 | 2.05 | 0.01 | 0.01 | 0.01 |
| SRR6442701 | 48 | 48 | 48 | 32 | 48 | 4.52 | 0.78 | 2.04 | 5.95 | 2 | 0.05 | 0.53 | 0.03 | 0.10 | 0.13 | 0.41 | 0.54 | 7742 | 0.41 | 0.22 | 0.23 | 0.95 | 2.05 | 0.01 | 0.03 | 0.03 |
| SRR6442702 | 42 | 42 | 42 | 22 | 42 | 4.87 | 0.79 | 1.76 | 4.35 | 3 | 0.03 | 0.47 | 0.04 | 0.07 | 0.07 | 0.28 | 0.49 | 19137 | 0.28 | 0.21 | 0.46 | 0.97 | 2.05 | 0.01 | 0.00 | 0.00 |
La función global nos da 26 medidas de diversidad que nos ayudan a entender la estructura de las comunidades microbianas. En general, estas medidas se dividen en riqueza, diversidad, dominancia, rareza, cobertura y uniformidad.
- El paquete
microbiomeofrece funciones para calcular cada uno de estos aspectos de las comunidades microbianas.
# Riqueza
tab <- richness(psd5)
# Dominancia
tab <- dominance(psd5, index = "all")
# Rareza
tab <- rarity(psd5, index = "all")
# Cobertura
tab <- coverage(psd5, threshold = 0.5)
# Desigualdad
tab <- inequality(psd5)
# Uniformidad
tab <- evenness(psd5, "all")- Veamos un ejemplo concreto estimando diversidad, graficando los resultados y calculando significancia estadística. Para esto usamos el paquete
ggpubrque genera “publication-ready plots”, algo que siempre es deseable (ejecutalibrary(ggpubr)).
# Generamos un objeto `phyloseq` sin taxa que sume 0 reads
psd5.2 <- prune_taxa(taxa_sums(psd5) > 0, psd5)
# Calculamos los índices de diversidad
tab <- diversities(psd5.2, index = "all")
# Y finalmente visualizamos la tabla de resultados
head(tab)## inverse_simpson gini_simpson shannon fisher coverage
## SRR6442697 6.948971 0.8560938 2.181249 4.100949 3
## SRR6442698 3.673899 0.7278096 1.913672 5.781418 2
## SRR6442699 6.461545 0.8452383 2.043740 3.117388 3
## SRR6442700 7.707637 0.8702586 2.412447 6.104153 3
## SRR6442701 4.520999 0.7788099 2.038536 5.949163 2
## SRR6442702 4.873575 0.7948118 1.759212 4.346443 3| inverse_simpson | gini_simpson | shannon | fisher | coverage | |
|---|---|---|---|---|---|
| SRR6442697 | 6.95 | 0.86 | 2.18 | 4.10 | 3 |
| SRR6442698 | 3.67 | 0.73 | 1.91 | 5.78 | 2 |
| SRR6442699 | 6.46 | 0.85 | 2.04 | 3.12 | 3 |
| SRR6442700 | 7.71 | 0.87 | 2.41 | 6.10 | 3 |
| SRR6442701 | 4.52 | 0.78 | 2.04 | 5.95 | 2 |
| SRR6442702 | 4.87 | 0.79 | 1.76 | 4.35 | 3 |
- Ahora necesitamos extraer la metadata de nuestro objeto
phyloseq.
psd5.2.meta <- meta(psd5.2)
head(psd5.2.meta)## sample_ID bioproject_accession study biosample_accession
## SRR6442697 EMA4 PRJNA428495 SRP128093 SAMN08292292
## SRR6442698 EMA3 PRJNA428495 SRP128093 SAMN08292291
## SRR6442699 EMA2 PRJNA428495 SRP128093 SAMN08292284
## SRR6442700 EMA19 PRJNA428495 SRP128093 SAMN08292283
## SRR6442701 EMA21 PRJNA428495 SRP128093 SAMN08292286
## SRR6442702 EMA20 PRJNA428495 SRP128093 SAMN08292285
## experiment run SRA_Sample geo_loc_name
## SRR6442697 SRX3533985 SRR6442697 SRS2809259 Chile: Estrecho_Magallanes
## SRR6442698 SRX3533984 SRR6442698 SRS2809258 Chile: Estrecho_Magallanes
## SRR6442699 SRX3533983 SRR6442699 SRS2809257 Chile: Estrecho_Magallanes
## SRR6442700 SRX3533982 SRR6442700 SRS2809256 Chile: Estrecho_Magallanes
## SRR6442701 SRX3533981 SRR6442701 SRS2809255 Chile: Estrecho_Magallanes
## SRR6442702 SRX3533980 SRR6442702 SRS2809254 Chile: Estrecho_Magallanes
## collection_date sample_type species
## SRR6442697 2017 skin Megaptera novaeangliae
## SRR6442698 2017 skin Megaptera novaeangliae
## SRR6442699 2017 skin Megaptera novaeangliae
## SRR6442700 2017 skin Megaptera novaeangliae
## SRR6442701 2017 skin Megaptera novaeangliae
## SRR6442702 2017 skin Megaptera novaeangliae
## common_name AvgSpotLen
## SRR6442697 humpback whale 501
## SRR6442698 humpback whale 500
## SRR6442699 humpback whale 501
## SRR6442700 humpback whale 500
## SRR6442701 humpback whale 499
## SRR6442702 humpback whale 500| sample_ID | bioproject_accession | study | biosample_accession | experiment | run | SRA_Sample | geo_loc_name | collection_date | sample_type | species | common_name | AvgSpotLen | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRR6442697 | EMA4 | PRJNA428495 | SRP128093 | SAMN08292292 | SRX3533985 | SRR6442697 | SRS2809259 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442698 | EMA3 | PRJNA428495 | SRP128093 | SAMN08292291 | SRX3533984 | SRR6442698 | SRS2809258 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442699 | EMA2 | PRJNA428495 | SRP128093 | SAMN08292284 | SRX3533983 | SRR6442699 | SRS2809257 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 501 |
| SRR6442700 | EMA19 | PRJNA428495 | SRP128093 | SAMN08292283 | SRX3533982 | SRR6442700 | SRS2809256 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
| SRR6442701 | EMA21 | PRJNA428495 | SRP128093 | SAMN08292286 | SRX3533981 | SRR6442701 | SRS2809255 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 499 |
| SRR6442702 | EMA20 | PRJNA428495 | SRP128093 | SAMN08292285 | SRX3533980 | SRR6442702 | SRS2809254 | Chile: Estrecho_Magallanes | 2017 | skin | Megaptera novaeangliae | humpback whale | 500 |
- Luego agregamos la tabla de diversidad a la metadata.
psd5.2.meta$Shannon <- tab$shannon
psd5.2.meta$InverseSimpson <- tab$inverse_simpson- En este ejercicio nos interesa comparar la diversidad entre especies de ballenas. Recordemos que tenemos datos para tres especies de ballenas: azul, fin y jorobada. Necesitamos crear una lista de comparasiones de a pares para poder visualizar y calcular significancia estadística de manera simultánea.
# Obtenemos las variables desde nuestro objeto `phyloseq`
spps <- levels(psd5.2.meta$species)
# Creamos una lista de lo que queremos comparar
pares.spps <- combn(seq_along(spps), 2, simplify = FALSE, FUN = function(i)spps[i])
# Imprimimos en pantalla el resultado
print(pares.spps)## [[1]]
## [1] "Balaenoptera musculus" "Balaenoptera physalus"
##
## [[2]]
## [1] "Balaenoptera musculus" "Megaptera novaeangliae"
##
## [[3]]
## [1] "Balaenoptera physalus" "Megaptera novaeangliae"- Con la función
ggviolinpodemos generar un gráfico de violín en un solo paso de la siguiente forma.
p1 <- ggviolin(psd5.2.meta, x = "species", y = "Shannon",
add = "boxplot", fill = "species", palette = c("#a6cee3", "#b2df8a", "#fdbf6f"))
print(p1)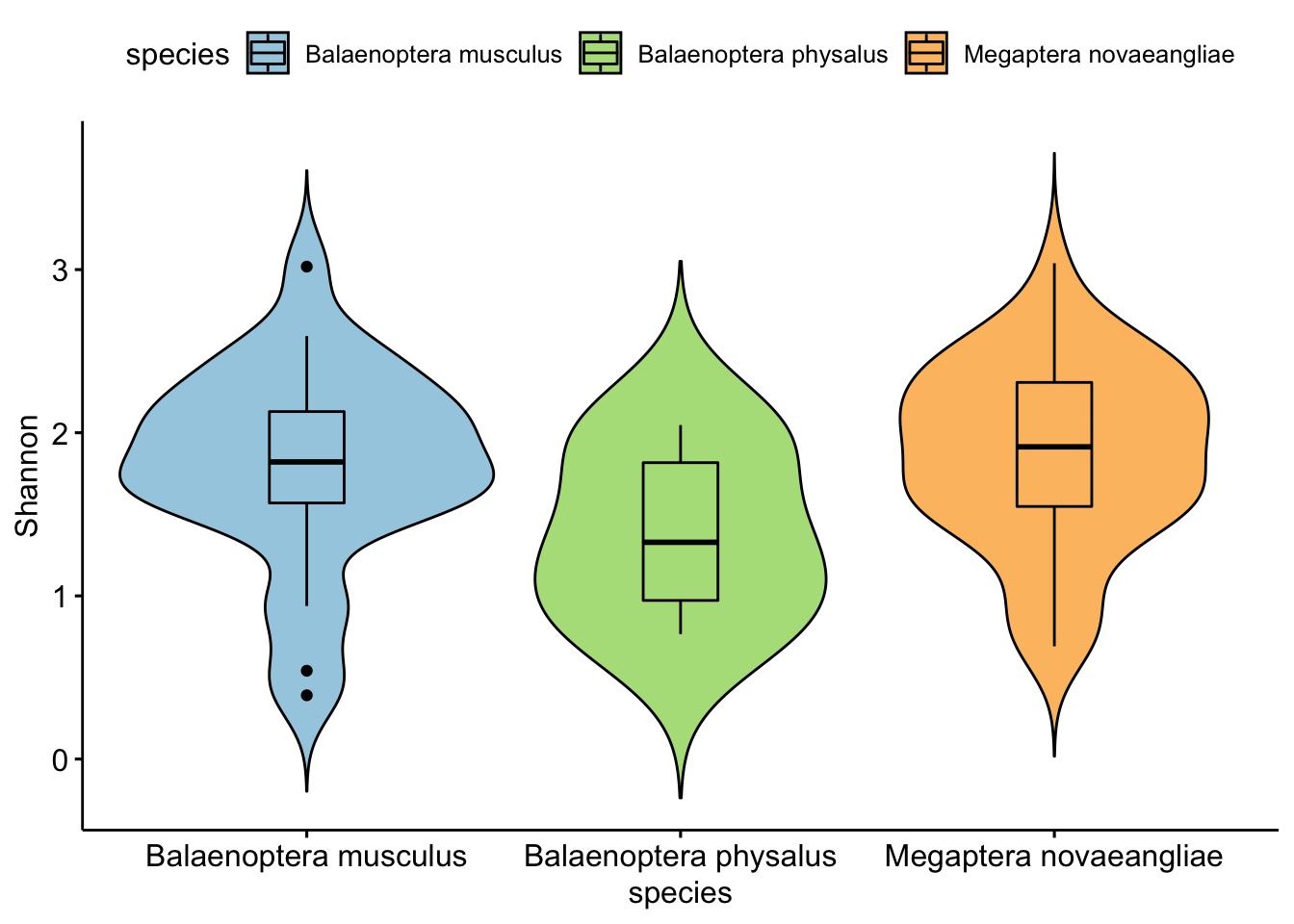
- Ahora necesitamos evaluar la significancia estadística entre los estimados de diversidad de las muestras de ballenas. De nuevo, en una línea, tenemos nuestra figura lista para el artículo.
p1 <- p1 + stat_compare_means(comparisons = pares.spps)
print(p1)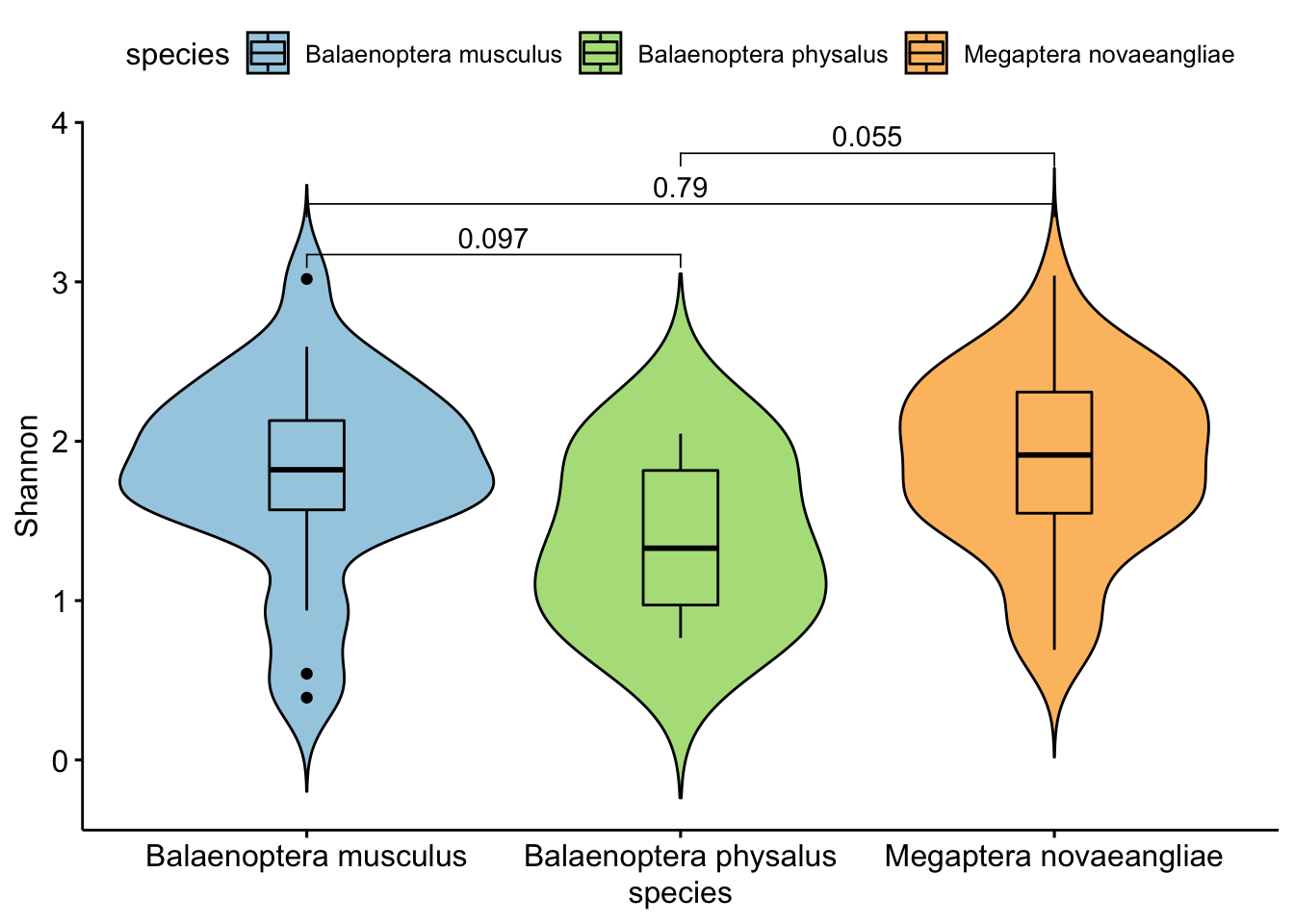
3.2 Diversidad beta y escalamiento multidimensional (Bray-Curtis, UniFrac, t-SNE)
En cuanto a diversidad beta podemos calcular similitud global a través de todas las muestras de interés o también podemos cuantificar la divergencia de un grupo y compararla con la divergencia de otro.
- Veamos este último caso primero.
# Calculamos las divergencias para ballena azul y jorobada
div.azul <- divergence(subset_samples(psd5, species == "Balaenoptera musculus"))
div.joro <- divergence(subset_samples(psd5, species == "Megaptera novaeangliae"))
# transformamos el resultado anterior en _dataframes_
data.frame(div.azul) -> df.div.azul
data.frame(div.joro) -> df.div.joro
# Agregamos columnas a nuestros _dataframes_
mutate(df.div.azul, species = "Blue Whale") -> df.div.azul
mutate(df.div.joro, species = "Humpback Whale") -> df.div.joro
# Cambiamos los nombres de las columans de manera que sean iguales an ambos _dataframes_
colnames(df.div.azul) <- c("divergence", "species")
colnames(df.div.joro) <- c("divergence", "species")
# Los combinamos en un _dataframe_
rbind(df.div.azul, df.div.joro) -> div.boxplot
# Y finalmente graficamos y realizamos una comparación estadística
p2 <- ggboxplot(data = div.boxplot, x = "species", y = "divergence", fill = "species", palette = c("#a6cee3", "#fdbf6f"))
p2 + stat_compare_means(comparisons = list(c("Blue Whale", "Humpback Whale")))
Existen diferentes medidas de similitud (o disimilitud, i.e., 1 - similitud) disponibles que nos permiten entender las relaciones entre nuestras muestras. En general todas producen matrices de distancia comparables. El paquete phyloseq ofrece un gran número de medidas de distancia. Las más populares son UniFrac y Weighted UniFrac (medidas que consideran filogenia) y otras independientes de filogenia como: Jaccard, Manhattan, Euclidian, Bray-Curtis, Canberra, etc. Por otra parte, la matriz de distancia resultante no se usa en aislación sino que en conjunto con algún método de ordinación o escalamiento multidimensional (ordination). De nuevo, phyloseq ofrece un gran número de métodos entre los cuales se encuentran: detrended y canonical correspondence analysis, Double Principal Coordinate Analysis, Non-metric MultiDimenstional Scaling, y MDS/PCoA.
- Probemos entonces hacer un análisis tipo PCoA con una matriz de distancia que considera las relaciones filogenéticas y otra que no.
psd5.mds.unifrac <- ordinate(psd5, method = "MDS", distance = "unifrac")
evals <- psd5.mds.unifrac$values$Eigenvalues
pord1 <- plot_ordination(psd5, psd5.mds.unifrac, color = "geo_loc_name") +
labs(col = "geo_loc_name") +
coord_fixed(sqrt(evals[2] / evals[1]))
psd5.mds.bray <- ordinate(psd5, method = "MDS", distance = "bray")
evals <- psd5.mds.bray$values$Eigenvalues
pord2 <- plot_ordination(psd5, psd5.mds.bray, color = "geo_loc_name") +
labs(col = "geo_loc_name") +
coord_fixed(sqrt(evals[2] / evals[1]))
grid.arrange(pord1, pord2)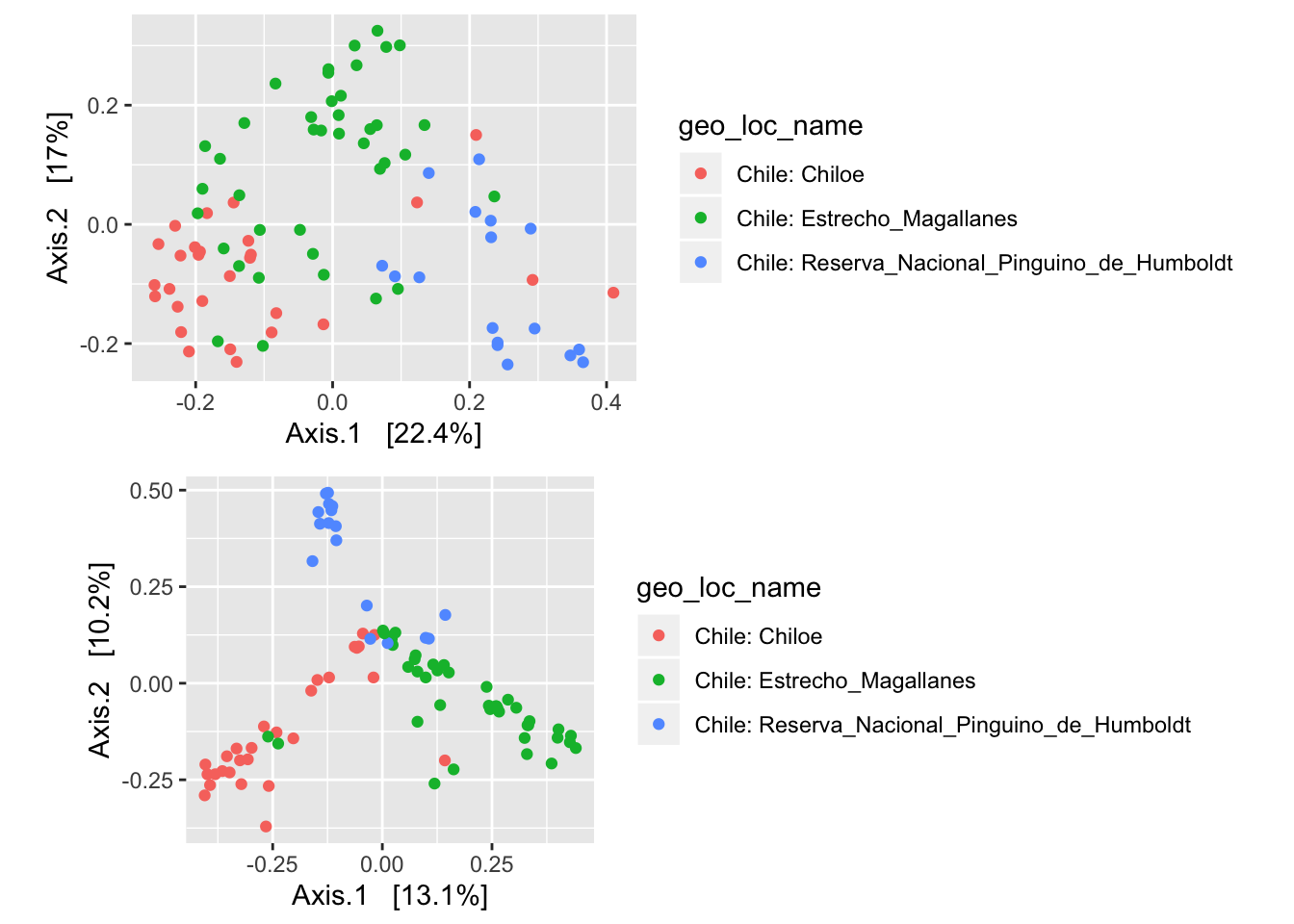
Nota que los gráficos de dispersión donde se visualiza este tipo de análisis están escalados según la cantidad de variación que los ejes explican. En general lo que estos métodos pretenden hacer es tratar de encontrar el menor número de vectores matemáticos que maximicen la separación entre las muestras (puntos en el gráfico). Esto nace de la imposibilidad de graficar eficientemente datos multidimencionales. Los datos que estamos analizando ciertamente son multidimensionales en el sentido que tenemos más de 100 taxa que varían simultáneamente en cada una de las 90+ muestras que tenemos. Volviendo a los ejes, estos no suman 100% porque hay otros ejes que no estamos usando para graficar y que contribuyen con el resto de la variación. Al graficarlos de manera simétrica distorsionaríamos las relaciones entre los puntos, especialmente si estamos comparando dos o más gráficos.
En específico para comunidades microbianas, el método de doble análisis de coordenadas principales o (DPCoA) es muy apropiado porque analiza conjuntamente dos tipos de datos: una tabla de disimilitud que representa diferencias entre especies y una matriz de abundancia que representa la distribución de especies entre las comunidades. El resultado final es un ensamble del espacio multidimensional que correlacionan las especies con las comunidades. El método fue publicado en 2004.
- Veamos un ejemplo con nuestros datos.
psd5.dpcoa.unifrac <- ordinate(psd5, method = "DPCoA", distance = "dpcoa")
evals <- psd5.dpcoa.unifrac$eig
pord3 <- plot_ordination(psd5, psd5.dpcoa.unifrac, color = "species", shape = "geo_loc_name") +
labs(col = "Species") +
coord_fixed(sqrt(evals[2] / evals[1])) +
scale_color_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f")) +
scale_fill_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f")) +
geom_point(size=4)
pord3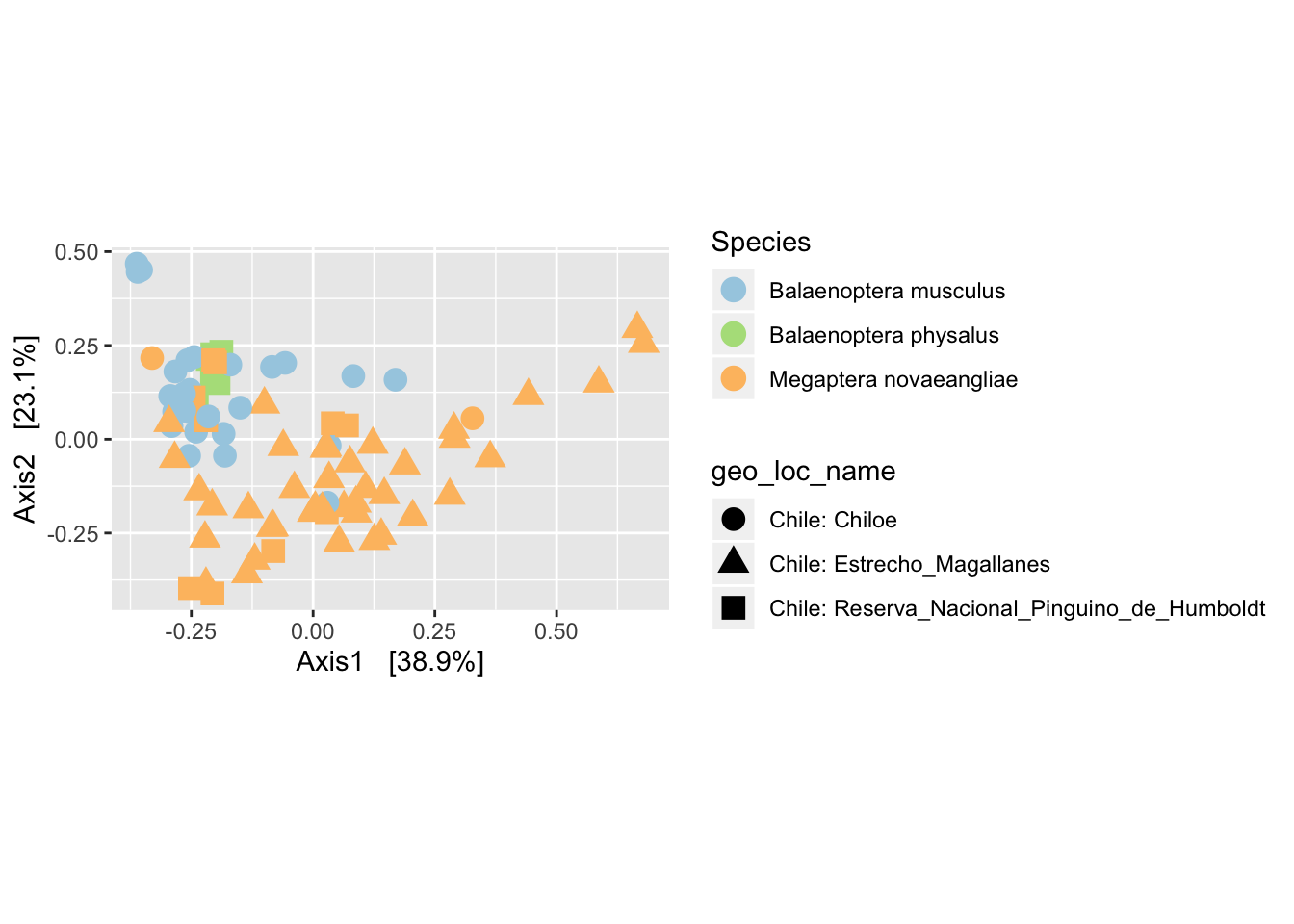
- Ahora exploremos escalamiento multidimensional usando un método reciente conocido como t-SNE o t-Distributed Stochastic Neighbor Embedding. t-SNE difiere de otros métodos en que hace énfasis en las distancias locales en vez de las distancias globales, de esta forma generando una mayor resolución o separación entre los puntos o muestras.
library(tsnemicrobiota)
tsne_res <- tsne_phyloseq(psd5, distance= "dpcoa", perplexity = 8, verbose=0, rng_seed = 3901)
# Graficamos
pord4 <- plot_tsne_phyloseq(psd5, tsne_res, color = "species", shape = "geo_loc_name") +
geom_point(size=4) +
scale_color_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f")) +
scale_fill_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f"))
grid.arrange(pord3, pord4)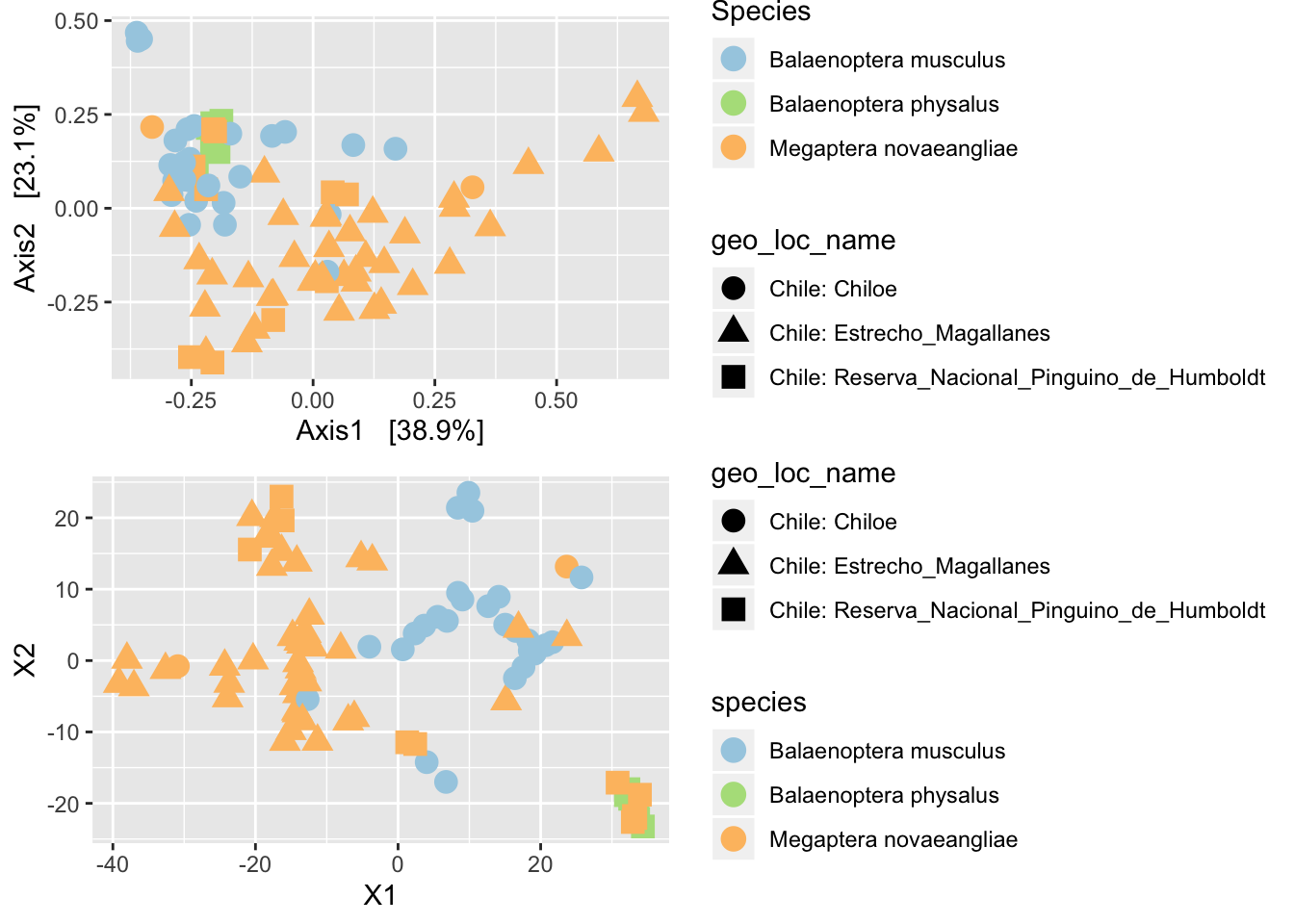
Ambos gráficos usan distintos métodos pero la misma medida de distancia. Los resultados son similares aunque las agrupaciones de puntos o muestras ocupan distinto espacio en el gráfico.
- Otro uso de estas medidas es a través de la visualización de la densidad de las muestras en el espacio.
method <- "tsne"
trans <- "hellinger"
distance <- "euclidean"
# Matriz de distancia
psd5 <- microbiome::transform(psd5, trans)
# Calculamos similitud entre muestras
dm <- vegdist(otu_table(psd5), distance)
# Corremos t-SNE
tsne_out <- Rtsne(dm, dims = 2, perplexity = 8)
proj <- tsne_out$Y
rownames(proj) <- rownames(otu_table(psd5))
data.frame(proj) -> proj
proj$species <- sample_data(psd5)[,11]
pland <- plot_landscape(proj[,1:2], legend = T, size = 4)
print(pland)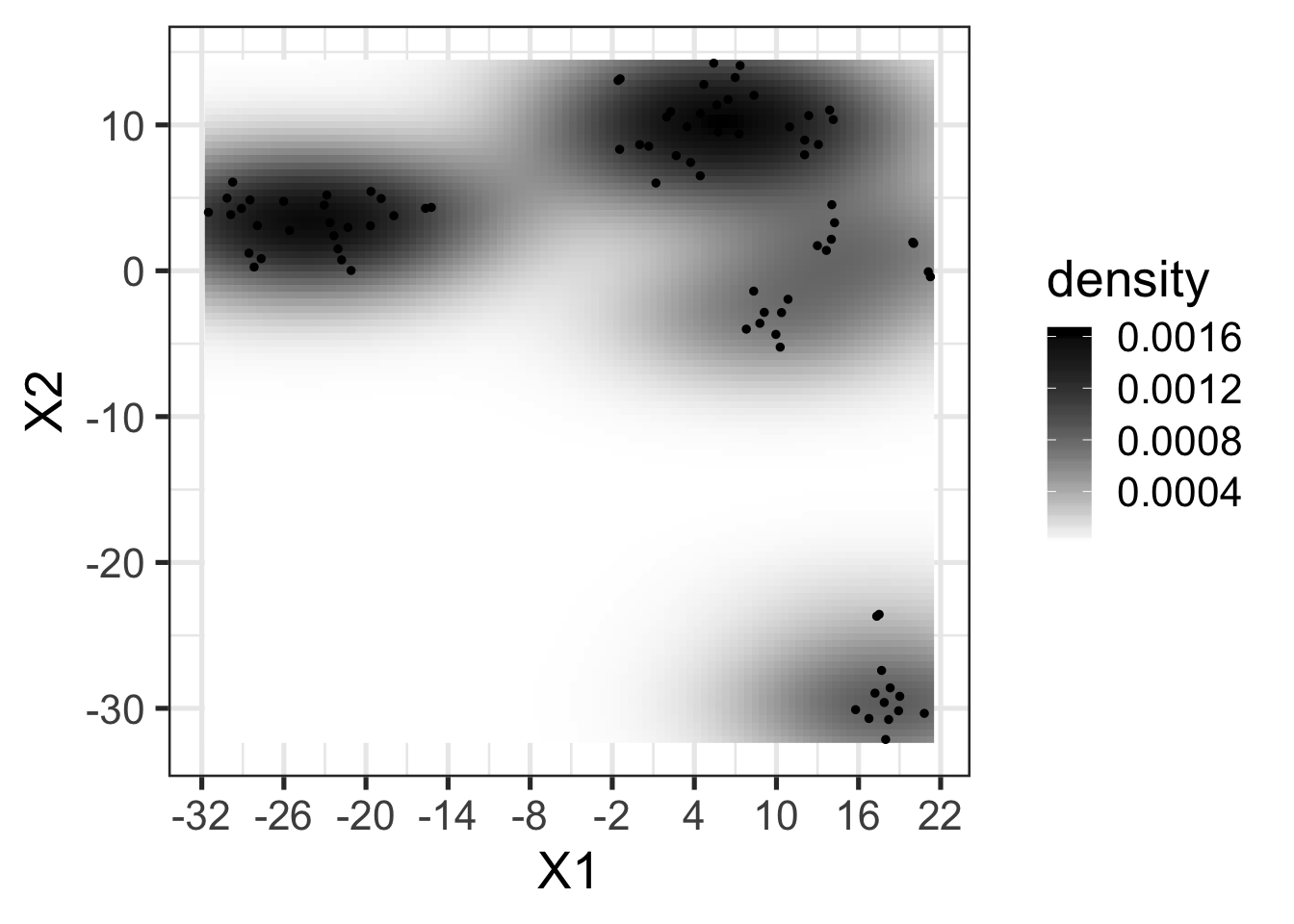
3.3 Análisis de abundancias y visualizaciones
3.3.1 Gráfico de barras apiladas
Una manera muy eficiente de obtener una visión general de la composición de las muestras es a través de un gráfico de barras apiladas. Existe una variadad de funciones que pueden hacer esto, sin embargo vamos a usar el paquete creado por un ex-miembro del laboratorio ya que tiene la ventaja de poder agrupar por hierarchical clustering las muestras entre otras ventajas.
# Necesitamos obtener las taxa más abundantes, en este caso el top 15
top15 <- get_top_taxa(physeq_obj = psd5, n = 15, relative = T,
discard_other = T, other_label = "Other")
# Ya que no todas las taxa fueron clasificadas a nivel de especie, generamos etiquetas compuestas de distintos rangos taxonómicos para el gráfico
top15 <- name_taxa(top15, label = "", species = F, other_label = "Other")
# Finalmente graficamos
fantaxtic_bar(top15, color_by = "Family", label_by = "Genus", facet_by = NULL, grid_by = NULL, other_color = "Grey") -> ptop15## Level N.color.shades Central.color
## 1 Cardiobacteriaceae 1 #6495ED
## 2 Flavobacteriaceae 1 #EDBC64
## 3 Moraxellaceae 5 #9A64ED
## 4 Gammaproteobacteria (Class) 2 #B7ED64
## 5 Xanthomonadaceae 1 #ED64DA
## 6 Enterobacteriaceae 2 #64ED77
## 7 Pseudomonadaceae 2 #ED6473
## 8 Burkholderiaceae 1 #64EDDEptop15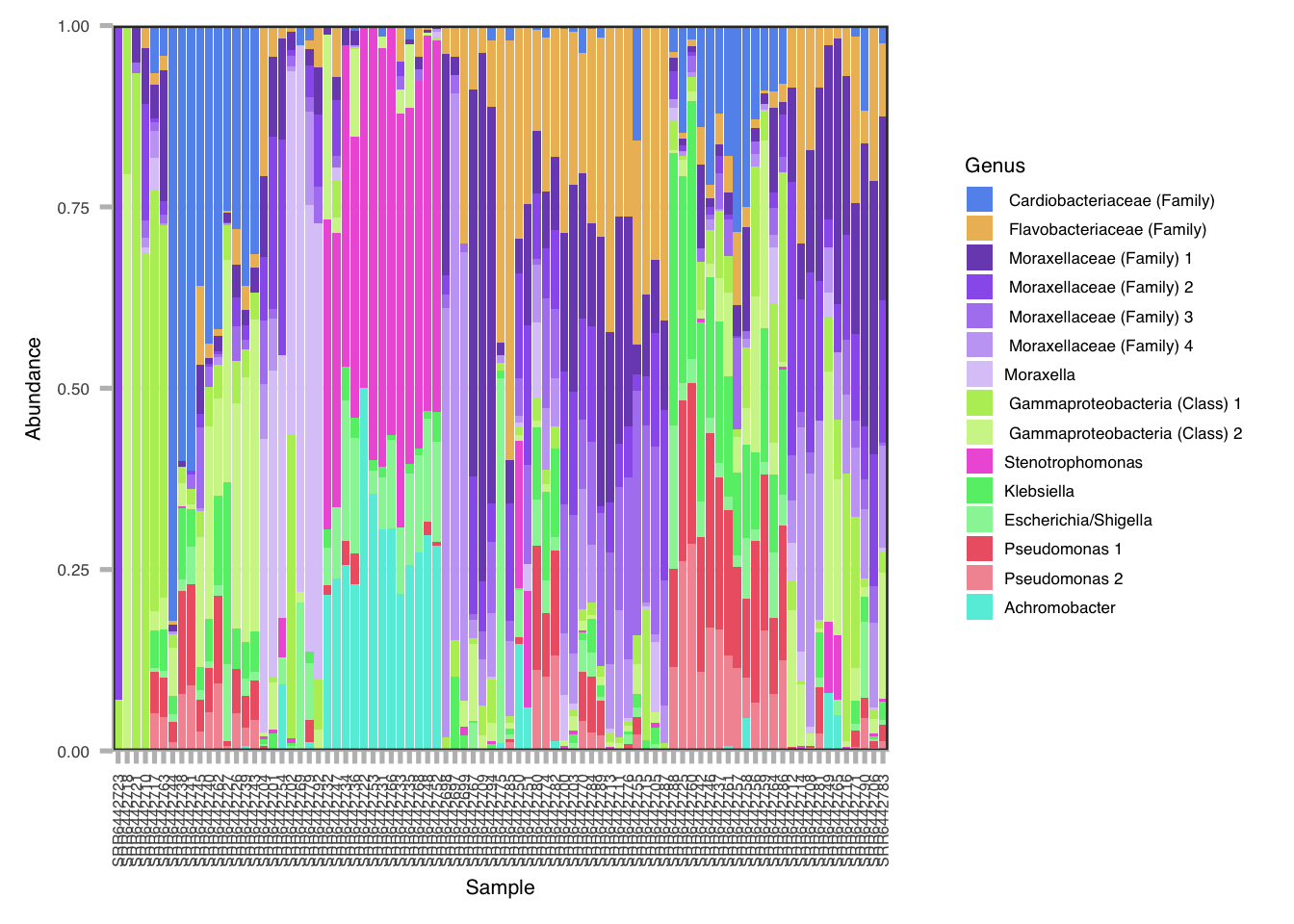
Podemos ver que existen patrones relativamente claros en el gráfico solamente a partir de los colores. Aunque tenemos el nombre de las muestras bajo cada barra, sería mejor poder parcelar este gráfico de manera que quede claro qué muestras corresponden a qué especie de ballena.
- La función
fantaxtic_barofrece estas posibilidades a través de los argumentosfacet_byygrid_by. Grafiquemos de nuevo.
fantaxtic_bar(top15, color_by = "Family", label_by = "Genus", facet_by = "species", grid_by = NULL, other_color = "Grey") -> ptop15.2## Level N.color.shades Central.color
## 1 Cardiobacteriaceae 1 #6495ED
## 2 Flavobacteriaceae 1 #EDBC64
## 3 Moraxellaceae 5 #9A64ED
## 4 Gammaproteobacteria (Class) 2 #B7ED64
## 5 Xanthomonadaceae 1 #ED64DA
## 6 Enterobacteriaceae 2 #64ED77
## 7 Pseudomonadaceae 2 #ED6473
## 8 Burkholderiaceae 1 #64EDDEptop15.2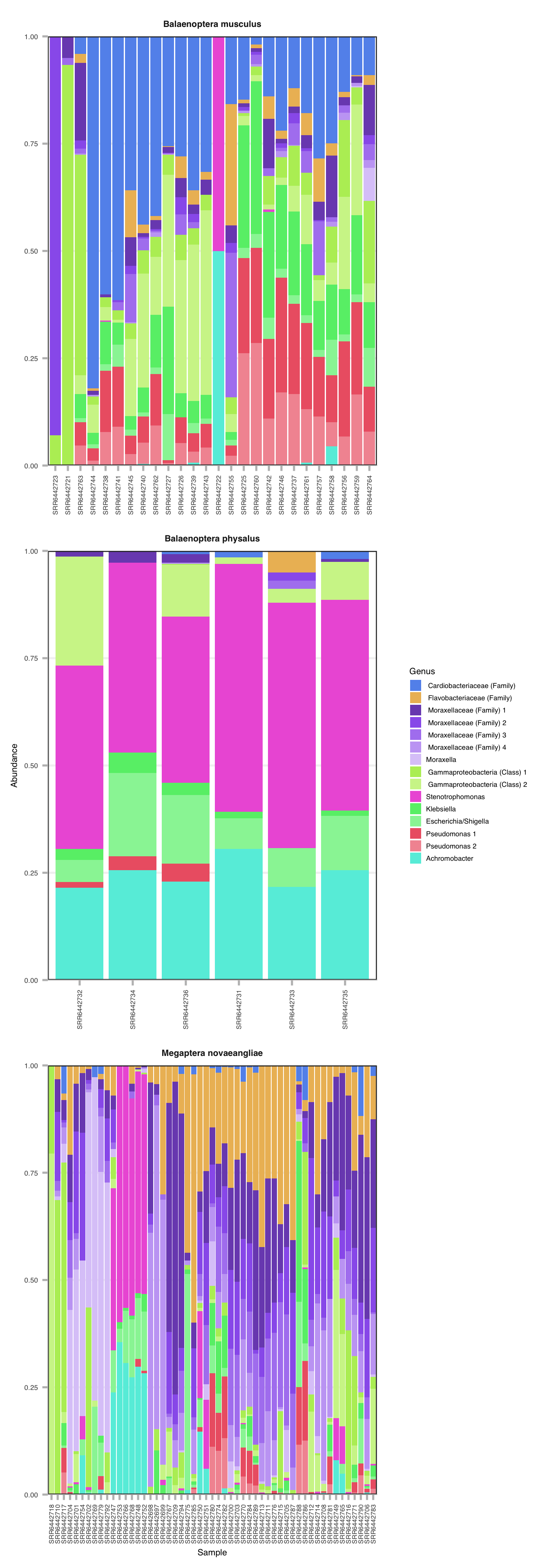
Ahora queda más claro y se puede observar que las distintas especies de ballena tienen un patrón similar entre sí que es diferente entre las otras, con algunas excepciones. Por ejemplo, para ballena jorobada podemos ver un conjunto de muestras que no se parecen al resto. Usando las herramientas ya aprendidas, ¿A qué corresponden esas muestras?
3.3.2 Diferentes visualizaciones de abundancias
Veamos ahora otras herramientas que nos permiten examinar la composición de estas comunidades microbianas. El paquete ampvis2, desarrollado por Mads Albertsen y Kasper Skytte Andersen, nos permite hacer precisamente esto. Primero transformemos el objeto phyloseq con el cual hemos estado trabajando en un objeto ampvis2.
library(ampvis2)
# Necesitamos extraer la tabla de read counts y la tabla de taxonomía del objeto psd5
# Generamos una copia para no sobreescribir psd5
obj <- psd5
# Cambiamos la orientación de la otu_table
t(otu_table(obj)) -> otu_table(obj)
# Extraemos las tablas
otutable <- data.frame(OTU = rownames(phyloseq::otu_table(obj)@.Data),
phyloseq::otu_table(obj)@.Data,
phyloseq::tax_table(obj)@.Data,
check.names = FALSE
)
# Extraemos la metadada
metadata <- data.frame(phyloseq::sample_data(obj),
check.names = FALSE
)
# ampvis2 requiere que 1) los rangos taxonómicos sean siete y vayan de Kingdom a Species y 2) la primera columna de la metadata sea el identificador de cada muestra
# Entonces duplicamos la columna Género y le cambiamos el nombre a Especie
otutable$Species = otutable$Genus
# Reordenamos la metadata
metadata <- metadata[,c("run","sample_ID","bioproject_accession","study","biosample_accession","experiment","SRA_Sample","geo_loc_name","collection_date","sample_type","species","common_name","AvgSpotLen")]
# finalmente generamos el objeto ampvis
av2 <- amp_load(otutable, metadata)- Ahora echemos un vistazo a la estructura de las comunidades utilizando “rank abundance curves”.
amp_rankabundance(av2, plot_log = T, group_by = "species")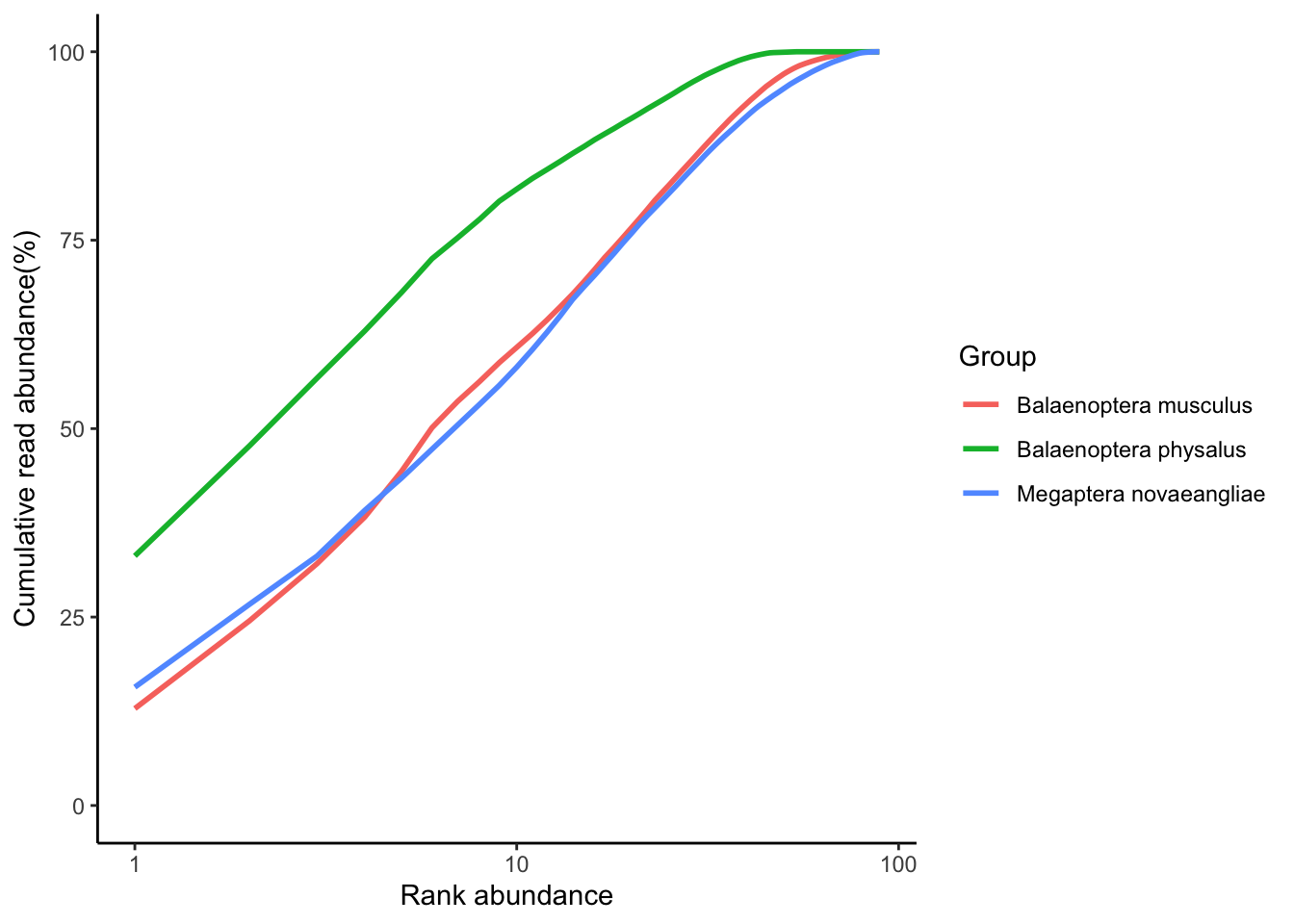
El gráfico nos muestra que en la medida que vamos sumando las taxa de mayor a menor abundancia (Rank Abundance) la abundancia de reads cumulativa va aumentando. Lo importante de observar es la forma de la curva. Una curva que sube rápidamente nos indica que las comunidades están dominadas por unas cuantas taxa. En cambio, lo que observamos en nuestros datos es que las taxa más abundantes solamente dan cuenta de aproximadamente el 25% de la abundancia cumulativa de reads.
- Veamos ahora qué taxa corresponde a ese 25%. Para esto podemos usar la función
amp_heatmap.
amp_heatmap(av2,
group_by = "species",
facet_by = "geo_loc_name",
plot_values = TRUE,
tax_show = 20,
tax_aggregate = "Genus",
tax_add = "Phylum",
plot_colorscale = "sqrt",
plot_legendbreaks = c(1, 5, 10)
)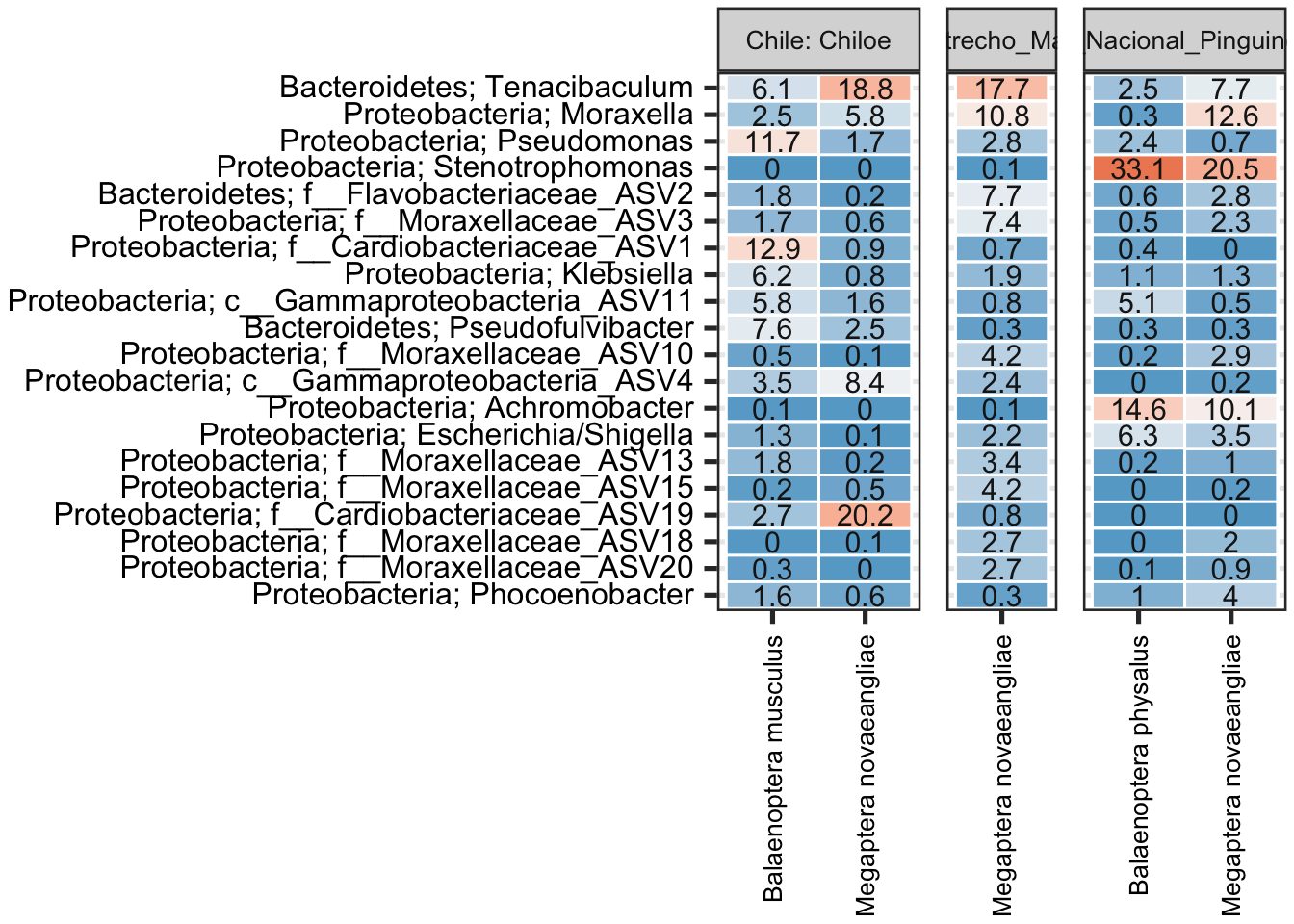
Tenacibaculum parece ser el género más abundante para todas las muestras en todas los sitios de muestreo con excepción de Balaenoptera physalus que está dominada por Stenotrphomonas. Moraxella y distintas variantes de Cardiobacteriaceae de género no conocido. Justamente estos resultados se ajustan a lo conocido para cetáceos y otros mamíferos marinos.
- También podemos realizar una visualización similar pero usando Box Plots.
amp_boxplot(av2,
group_by = "species",
tax_show = 20,
tax_aggregate = "Genus",
tax_add = "Phylum",
adjust_zero = T,
plot_log = T) +
scale_color_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f")) +
scale_fill_manual(values = c("#a6cee3", "#b2df8a", "#fdbf6f"))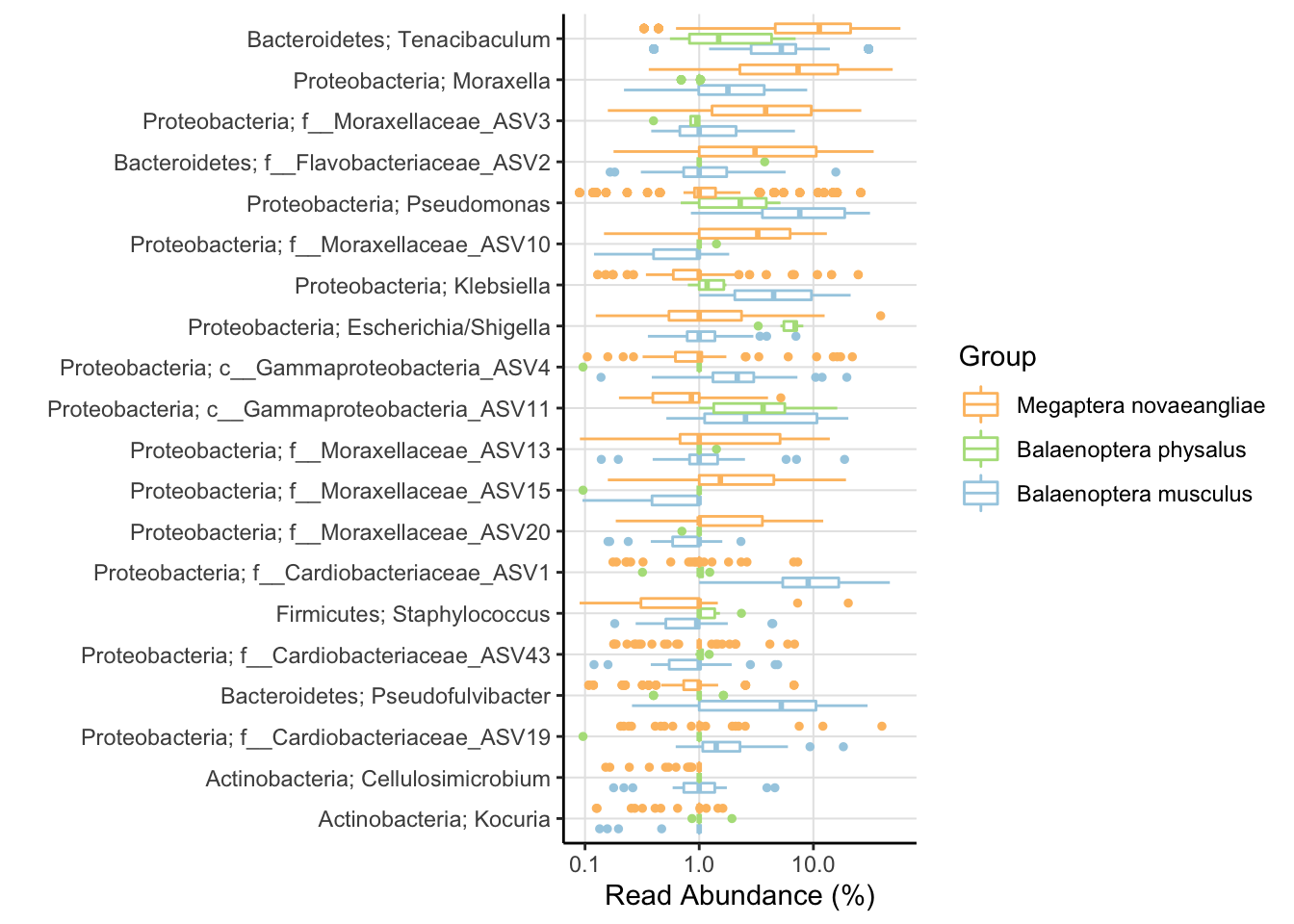
- Veamos ahora si es que algunos de estos microorganismos están compartidos entre todas las muestras. Para esto debemos calcular el core microbiome o el conjunto de taxa compartidas entre un cierto umbral porcentual de muestras y de prevalencia intra-muestra.
amp_core(av2,
tax_aggregate = "Family",
group_by = "Sample",
abund_thrh = 0.5)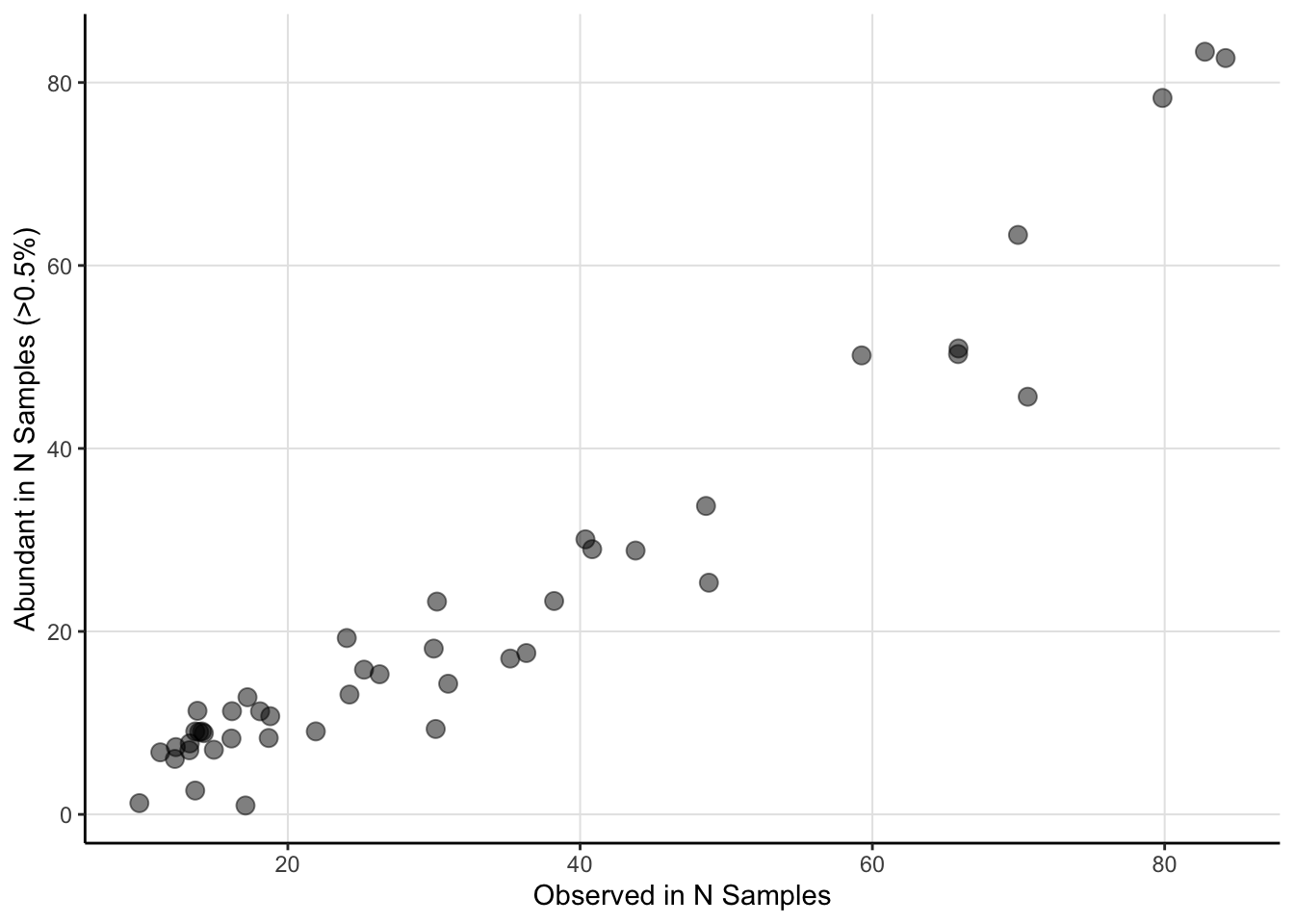
- Y visto de otra manera en un diagrama de Venn.
amp_venn(av2, group_by = "species", cut_a = 0, cut_f = 50, text_size = 3)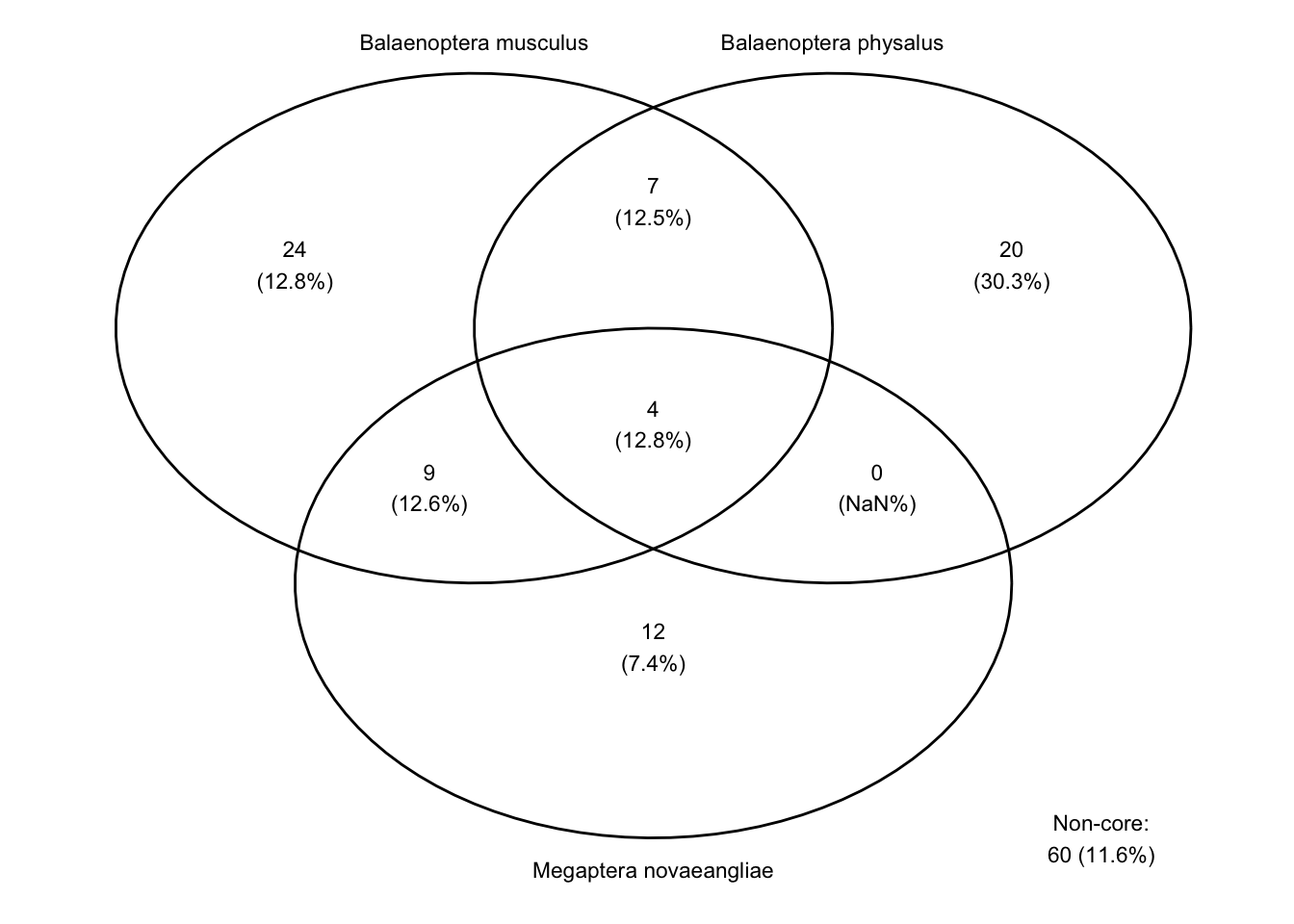
3.3.3 Análisis de abundancia diferencial de microorganismos
Hasta ahora hemos visto principalmente análisis exploratorios y algunos test estadísticos para diversidad alfa y beta. Sin embargo, muchas veces queremos determinar exactamente qué taxa está más representada en una condición versus otra y en qué medida. El procedimiento es análogo al análisis de expresión diferencial de genes en transcritómica, e.g., RNA-seq. Es tan así que justamente ocupamos uno de los paquetes de R más populares en transcriptómica, DESeq2. Ahora, nuestras muestras se secuenciaron al mismo tiempo y se intentó que se produjera una profundidad uniforme a través de todas las muestras. En la práctica esto no ocurre y al momento de analizar las muestras en el contexto del análisis diferencial de abundancia debemos corregir por dos situaciones: el tamaño desigual de las muestras (en número de reads) y la correlación de la variabilidad de los datos con la media de éstos. Para esto último, utilizamos una transformación llamada Variance Stabilizing Transformation, cuyo objetivo es encontrar una función ƒ que se aplique a los valores de read counts, de tal forma que en los nuevos valores y = ƒ(x), la variabilidad de y no se relacione con sus valores medios.
- Comencemos por aplicar la transformación en los datos.
# Creamos un objeto DESeq2 con la función `phyloseq_to_deseq2`
diagdds = phyloseq_to_deseq2(psd5, ~species)
# Calculamos los factores de tamaño como parte de la normalización de las muestras
# calculate geometric means prior to estimate size factors
gm_mean = function(x, na.rm=TRUE){
exp(sum(log(x[x > 0]), na.rm=na.rm) / length(x))
}
geoMeans = apply(counts(diagdds), 1, gm_mean)
diagdds = estimateSizeFactors(diagdds, geoMeans = geoMeans)
# Normalizamos y realizamos el test paramétrico de Wald para determinar taxa diferencialmente abundante.
diagdds = DESeq(diagdds, test="Wald", fitType="local")Hasta ahora hemos transformado nuestro objeto phyloseq en un objeto DESeq2 de nombre diagdds, y hemos normalizado las cuentas y realizado un test paramétrico (Wald Test).
- Nos queda entonces revisar los resultados usando la función
results.
# Guardamos los resultados en el objeto res
res = results(diagdds, cooksCutoff = FALSE)
# hacemos un poco de aseo y ordenamos la tabla de resultados según p-value, y dejamos los valores NA al final
res = res[order(res$padj, na.last=NA), ]- Ahora nosotros queremos averiguar sobre ciertos contrastes específicos entre condiciones, e.g., ballena jorobada versus ballena azul. En el contraste, pasamos un vector con el nombre de la columna en la metadata (“species”) e indicamos el numerador de la comparación (“Megaptera novaeangliae”) y el denominador (“Balaenoptera musculus”). Por lo tanto el
log2FoldChangepositivo indicará que ese microorganismo es más abundante en ballena jorobada que en ballena azul, y viceversa.
res.joro.azul <- results(diagdds, contrast=c("species","Megaptera novaeangliae","Balaenoptera musculus"))Descarga el archivo
res.joro.azul.RDSAQUÍVeamos qué hay en
res.joro.azul
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| ASV1 | 2719.4817399 | -7.1224534 | 1.0359380 | -6.8753665 | 0.0000000 | 0.0000000 |
| ASV2 | 2022.5405509 | 5.4006313 | 0.7461453 | 7.2380427 | 0.0000000 | 0.0000000 |
| ASV3 | 2100.9914000 | 5.3802668 | 0.6766070 | 7.9518346 | 0.0000000 | 0.0000000 |
| ASV4 | 489.6362991 | 0.8724453 | 0.8271381 | 1.0547760 | 0.2915278 | 0.3442805 |
| ASV5 | 1138.1509390 | 13.0147565 | 1.3019415 | 9.9964222 | 0.0000000 | 0.0000000 |
| ASV6 | 45.6810800 | 5.5840818 | 1.8653474 | 2.9935880 | 0.0027572 | 0.0049735 |
| ASV7 | 245.1490808 | 4.4924803 | 0.9791466 | 4.5881589 | 0.0000045 | 0.0000168 |
| ASV8 | 1211.9442253 | -3.2250402 | 0.9315225 | -3.4621172 | 0.0005359 | 0.0012307 |
| ASV9 | 148.8626474 | 4.0117257 | 0.8333867 | 4.8137625 | NA | NA |
| ASV10 | 548.7130326 | 6.4054200 | 0.7366579 | 8.6952434 | 0.0000000 | 0.0000000 |
| ASV11 | 1009.5925018 | -5.0854806 | 0.8255799 | -6.1598889 | 0.0000000 | 0.0000000 |
| ASV12 | 658.7876904 | -3.2447714 | 1.0411077 | -3.1166530 | 0.0018292 | 0.0034895 |
| ASV13 | 410.6582559 | 3.0099935 | 0.8716746 | 3.4531160 | 0.0005542 | 0.0012494 |
| ASV14 | 140.0717828 | 3.8198888 | 1.0080979 | 3.7892044 | NA | NA |
| ASV15 | 379.3194323 | 7.5644546 | 0.8161418 | 9.2685548 | 0.0000000 | 0.0000000 |
| ASV16 | 285.7620345 | 7.6440491 | 0.9097626 | 8.4022455 | 0.0000000 | 0.0000000 |
| ASV17 | 379.4487960 | 14.2183560 | 1.4003173 | 10.1536672 | 0.0000000 | 0.0000000 |
| ASV18 | 203.0595008 | 9.1750665 | 1.1303913 | 8.1167175 | 0.0000000 | 0.0000000 |
| ASV19 | 69.8158128 | -2.2614994 | 0.9908843 | -2.2823041 | 0.0224714 | 0.0331721 |
| ASV20 | 299.9612618 | 5.6657701 | 0.9446883 | 5.9975021 | 0.0000000 | 0.0000000 |
| ASV21 | 91.1653741 | 7.8577937 | 1.1458691 | 6.8574966 | 0.0000000 | 0.0000000 |
| ASV22 | 522.4807634 | -3.8585579 | 1.1993634 | -3.2171718 | 0.0012946 | 0.0027209 |
| ASV23 | 100.3448777 | 10.0985821 | 1.3550063 | 7.4527934 | 0.0000000 | 0.0000000 |
| ASV24 | 345.5275036 | 8.1857232 | 1.3845014 | 5.9123979 | 0.0000000 | 0.0000000 |
| ASV25 | 278.9908685 | -2.5125876 | 1.5875681 | -1.5826644 | 0.1134980 | 0.1436097 |
| ASV26 | 62.1557348 | -0.4907744 | 1.4756983 | -0.3325709 | 0.7394582 | 0.7904553 |
| ASV27 | 189.6100970 | -3.6035868 | 1.2050796 | -2.9903310 | 0.0027868 | 0.0049735 |
| ASV28 | 218.1259662 | -0.4960691 | 0.8838224 | -0.5612769 | 0.5746088 | 0.6361740 |
| ASV29 | 348.5666726 | -9.2693864 | 1.2067447 | -7.6813154 | 0.0000000 | 0.0000000 |
| ASV30 | 19.9949713 | -6.9404408 | 2.0012541 | -3.4680458 | NA | NA |
| ASV31 | 9.4574754 | 4.4204992 | 1.8265012 | 2.4202006 | 0.0155119 | 0.0234571 |
| ASV32 | 135.9042193 | 4.6452237 | 1.0957355 | 4.2393659 | 0.0000224 | 0.0000678 |
| ASV33 | 12.2150782 | 3.8067681 | 1.2271358 | 3.1021570 | NA | NA |
| ASV34 | 14.5758192 | 6.9563749 | 0.9863495 | 7.0526474 | 0.0000000 | 0.0000000 |
| ASV35 | 78.7642402 | 1.0289747 | 0.8831016 | 1.1651827 | NA | NA |
| ASV36 | 15.6844519 | 4.4745682 | 1.5457245 | 2.8948031 | 0.0037940 | 0.0065340 |
| ASV37 | 104.0931206 | 6.3198854 | 1.0890983 | 5.8028606 | 0.0000000 | 0.0000000 |
| ASV38 | 63.0999441 | 7.0198666 | 1.2124308 | 5.7899111 | 0.0000000 | 0.0000000 |
| ASV39 | 71.6857366 | -4.1210982 | 0.9699437 | -4.2488015 | 0.0000215 | 0.0000666 |
| ASV40 | 11.7563573 | 4.6362327 | 1.8491255 | 2.5072570 | NA | NA |
| ASV41 | 63.6924912 | 6.7765578 | 1.5717381 | 4.3115057 | 0.0000162 | 0.0000522 |
| ASV42 | 39.8228286 | 3.9641573 | 1.1704109 | 3.3869791 | 0.0007067 | 0.0015108 |
| ASV43 | 29.6927766 | 0.6482185 | 0.9863497 | 0.6571893 | 0.5110592 | 0.5761031 |
| ASV44 | 4.6851145 | -4.9032274 | 1.1881480 | -4.1267818 | 0.0000368 | 0.0001061 |
| ASV45 | 31.3542359 | -5.8903163 | 1.0674036 | -5.5183589 | 0.0000000 | 0.0000001 |
| ASV46 | 52.3468950 | 8.1940044 | 1.2045392 | 6.8026052 | 0.0000000 | 0.0000000 |
| ASV47 | 24.3460080 | 5.1994788 | 1.1498081 | 4.5220406 | 0.0000061 | 0.0000223 |
| ASV48 | 9.5531988 | -3.1476568 | 1.9364467 | -1.6254807 | NA | NA |
| ASV49 | 28.1689872 | 4.3313913 | 1.3563693 | 3.1933718 | 0.0014062 | 0.0028817 |
| ASV50 | 89.6234760 | -8.8288920 | 1.2859692 | -6.8655549 | 0.0000000 | 0.0000000 |
| ASV51 | 42.8236021 | 7.0692754 | 1.5947984 | 4.4327079 | 0.0000093 | 0.0000328 |
| ASV52 | 1.0011767 | 2.5999491 | 1.5148878 | 1.7162651 | 0.0861135 | 0.1112300 |
| ASV53 | 33.2080947 | 1.4605265 | 0.9766925 | 1.4953800 | 0.1348153 | 0.1655158 |
| ASV54 | 82.8130820 | -2.1815886 | 1.1338739 | -1.9240134 | 0.0543529 | 0.0732583 |
| ASV55 | 9.1745027 | 3.2672435 | 1.9477963 | 1.6774051 | 0.0934633 | 0.1194789 |
| ASV56 | 11.3403634 | 4.6971790 | 1.4994164 | 3.1326715 | 0.0017322 | 0.0034095 |
| ASV57 | 16.5241450 | 1.8610673 | 1.5605106 | 1.1926015 | 0.2330255 | 0.2778381 |
| ASV58 | 13.2662521 | 2.8954770 | 1.4432286 | 2.0062497 | 0.0448296 | 0.0624592 |
| ASV59 | 99.5242996 | -10.6308795 | 1.2924338 | -8.2254731 | 0.0000000 | 0.0000000 |
| ASV60 | 68.5859180 | 1.1857152 | 1.2128028 | 0.9776653 | 0.3282399 | 0.3839788 |
| ASV61 | 2.5896101 | 0.5237964 | 0.9994077 | 0.5241068 | NA | NA |
| ASV62 | 7.2983417 | 4.2913582 | 1.4469920 | 2.9657096 | 0.0030199 | 0.0052741 |
| ASV63 | 10.6361249 | 7.2294633 | 1.8091772 | 3.9959952 | 0.0000644 | 0.0001816 |
| ASV64 | 83.5838226 | -2.6340549 | 1.2610396 | -2.0887963 | 0.0367261 | 0.0517504 |
| ASV65 | 47.6516393 | 10.1206302 | 1.4127059 | 7.1640036 | 0.0000000 | 0.0000000 |
| ASV66 | 70.2942412 | 5.2089523 | 1.4217267 | 3.6638212 | 0.0002485 | 0.0006032 |
| ASV67 | 24.7841554 | 5.1537365 | 1.2188111 | 4.2284950 | 0.0000235 | 0.0000695 |
| ASV68 | 14.2934620 | 5.5087333 | 1.4218510 | 3.8743394 | 0.0001069 | 0.0002821 |
| ASV69 | 21.2132029 | 1.7036268 | 1.1444604 | 1.4885853 | 0.1365966 | 0.1660586 |
| ASV70 | 68.7558900 | -0.3460662 | 1.6848409 | -0.2053999 | 0.8372597 | 0.8516169 |
| ASV71 | 6.2303015 | -0.2601080 | 1.2445510 | -0.2089974 | NA | NA |
| ASV72 | 14.6638119 | 7.0648004 | 1.6396906 | 4.3086180 | 0.0000164 | 0.0000522 |
| ASV73 | 7.5897409 | 5.3428616 | 1.4510121 | 3.6821619 | 0.0002313 | 0.0005735 |
| ASV74 | 5.0789582 | -1.3853182 | 1.1394033 | -1.2158278 | NA | NA |
| ASV75 | 19.8697343 | 24.2907265 | 1.8316754 | 13.2614802 | 0.0000000 | 0.0000000 |
| ASV76 | 1.2239068 | -1.9354764 | 1.2311449 | -1.5720947 | 0.1159286 | 0.1437514 |
| ASV77 | 9.0374513 | -9.8994430 | 1.4981683 | -6.6076975 | 0.0000000 | 0.0000000 |
| ASV78 | 21.1929292 | 8.8483467 | 1.6584029 | 5.3354626 | 0.0000001 | 0.0000004 |
| ASV79 | 13.7751655 | -2.6546117 | 1.4029815 | -1.8921217 | 0.0584748 | 0.0771369 |
| ASV80 | 28.3678395 | 23.0655138 | 1.6147380 | 14.2843691 | 0.0000000 | 0.0000000 |
| ASV81 | 9.9644103 | -2.8946247 | 0.7983325 | -3.6258383 | NA | NA |
| ASV82 | 5.5165903 | 5.0158379 | 1.8121294 | 2.7679247 | 0.0056414 | 0.0095827 |
| ASV83 | 22.0290096 | 7.8115706 | 1.2982118 | 6.0171773 | 0.0000000 | 0.0000000 |
| ASV84 | 5.3880867 | 3.5536543 | 1.1892872 | 2.9880540 | 0.0028076 | 0.0049735 |
| ASV85 | 1.7369590 | -3.9773770 | 1.3253471 | -3.0010078 | 0.0026909 | 0.0049735 |
| ASV86 | 8.6940194 | 0.9909398 | 1.7309680 | 0.5724772 | 0.5669987 | 0.6334040 |
| ASV87 | 2.6463367 | -4.9527209 | 1.2607597 | -3.9283623 | 0.0000855 | 0.0002305 |
| ASV88 | 22.4846884 | -7.9573124 | 1.0142627 | -7.8454155 | 0.0000000 | 0.0000000 |
| ASV89 | 29.2974383 | -5.7553117 | 1.0928471 | -5.2663465 | 0.0000001 | 0.0000005 |
| ASV90 | 5.5849388 | 4.5854927 | 2.0669960 | 2.2184332 | 0.0265253 | 0.0386957 |
| ASV91 | 16.5698452 | -7.4960519 | 1.3749392 | -5.4519154 | 0.0000000 | 0.0000002 |
| ASV92 | 22.6522435 | -4.9676859 | 1.2977662 | -3.8278742 | 0.0001293 | 0.0003339 |
| ASV93 | 6.4062042 | -4.0851822 | 1.5797024 | -2.5860455 | 0.0097084 | 0.0156343 |
| ASV94 | 1.7970262 | 3.0983231 | 1.2328023 | 2.5132359 | 0.0119629 | 0.0190180 |
| ASV95 | 11.9501380 | -7.6996523 | 1.3118837 | -5.8691576 | 0.0000000 | 0.0000000 |
| ASV96 | 2.8351525 | 0.3828029 | 1.6800387 | 0.2278536 | 0.8197600 | 0.8470854 |
| ASV97 | 3.7248506 | 5.1135350 | 1.5097237 | 3.3870667 | 0.0007064 | 0.0015108 |
| ASV98 | 4.3233609 | 4.8042120 | 1.9622148 | 2.4483619 | 0.0143507 | 0.0219690 |
| ASV99 | 2.8243846 | 3.5024621 | 1.0975926 | 3.1910403 | 0.0014176 | 0.0028817 |
| ASV100 | 9.9778511 | -4.6282742 | 1.2648220 | -3.6592297 | 0.0002530 | 0.0006032 |
| ASV101 | 7.1881927 | -0.4163206 | 1.6084949 | -0.2588262 | 0.7957694 | 0.8292050 |
| ASV102 | 8.2413114 | -3.7473291 | 1.1989296 | -3.1255621 | 0.0017747 | 0.0034384 |
| ASV103 | 8.3102765 | -6.7562778 | 1.5258751 | -4.4278053 | 0.0000095 | 0.0000328 |
| ASV104 | 6.7857308 | 5.7064612 | 1.6741681 | 3.4085353 | 0.0006531 | 0.0014462 |
| ASV105 | 5.0488641 | 4.1346140 | 1.5935323 | 2.5946219 | 0.0094695 | 0.0154502 |
| ASV106 | 0.9541234 | -1.1028164 | 2.0346151 | -0.5420270 | 0.5877999 | 0.6450193 |
| ASV107 | 4.1007567 | 4.9980913 | 1.1373183 | 4.3946286 | 0.0000111 | 0.0000372 |
| ASV108 | 5.4038096 | -3.6271094 | 1.2133551 | -2.9893224 | 0.0027960 | 0.0049735 |
| ASV109 | 1.3501927 | -0.4206071 | 1.4415801 | -0.2917681 | 0.7704639 | 0.8096400 |
| ASV110 | 2.7371281 | 3.3005287 | 1.7374702 | 1.8996174 | 0.0574833 | 0.0766445 |
| ASV111 | 2.4259080 | 4.5000589 | 1.8203672 | 2.4720611 | 0.0134337 | 0.0210857 |
| ASV112 | 1.5325388 | -0.6703858 | 1.9366425 | -0.3461588 | 0.7292234 | 0.7862930 |
| ASV113 | 5.1286581 | 5.5651234 | 1.5523269 | 3.5850202 | 0.0003371 | 0.0007886 |
| ASV114 | 3.2084055 | 4.2076848 | 1.5538169 | 2.7079669 | 0.0067697 | 0.0113438 |
| ASV115 | 1.8693006 | 4.1459636 | 1.5967342 | 2.5965270 | 0.0094171 | 0.0154502 |
| ASV116 | 7.7258899 | -6.1720272 | 1.5544558 | -3.9705388 | 0.0000717 | 0.0001976 |
| ASV117 | 4.6721853 | -6.3099610 | 1.6600099 | -3.8011586 | 0.0001440 | 0.0003645 |
| ASV118 | 0.9186556 | -1.6434260 | 1.7179194 | -0.9566375 | 0.3387503 | 0.3925704 |
| ASV119 | 7.0228498 | -1.8945403 | 0.8629677 | -2.1953780 | 0.0281365 | 0.0401026 |
| ASV120 | 0.8432035 | 0.1593532 | 1.9188207 | 0.0830475 | 0.9338138 | 0.9338138 |
| ASV121 | 3.3482616 | -2.7839431 | 1.2669338 | -2.1973864 | 0.0279929 | 0.0401026 |
| ASV122 | 3.6359471 | -4.5928491 | 1.8643476 | -2.4635155 | 0.0137582 | 0.0213252 |
| ASV123 | 1.7902754 | -1.4023022 | 1.5327540 | -0.9148906 | 0.3602491 | 0.4136193 |
| ASV124 | 2.6697932 | 2.1839877 | 1.3857261 | 1.5760602 | 0.1150119 | 0.1437514 |
| ASV125 | 1.1415391 | -0.7290996 | 1.8509376 | -0.3939082 | 0.6936488 | 0.7544952 |
| ASV126 | 2.9465902 | -1.8431157 | 1.2640321 | -1.4581241 | 0.1448064 | 0.1743300 |
| ASV127 | 2.9109028 | 0.9547596 | 1.1013290 | 0.8669159 | 0.3859881 | 0.4391057 |
| ASV128 | 2.9624475 | -4.1618781 | 1.3116973 | -3.1728951 | 0.0015093 | 0.0030185 |
| ASV129 | 2.1092384 | -0.3088813 | 1.5096505 | -0.2046045 | 0.8378811 | 0.8516169 |
| ASV130 | 1.4749075 | -0.2884521 | 0.9376518 | -0.3076324 | 0.7583621 | 0.8037342 |
| ASV131 | 2.3920165 | -2.4765750 | 1.3681047 | -1.8102232 | 0.0702612 | 0.0917093 |
| ASV132 | 1.3600064 | 0.1650124 | 1.5324853 | 0.1076763 | 0.9142524 | 0.9216854 |
| ASV133 | 0.7154383 | 2.8063290 | 1.4139574 | 1.9847338 | 0.0471741 | 0.0649954 |
| ASV134 | 1.0638791 | -1.5182223 | 1.3665996 | -1.1109489 | NA | NA |
| ASV135 | 1.4368076 | -4.0800926 | 1.6943303 | -2.4080857 | 0.0160364 | 0.0239580 |
| ASV136 | 0.6620716 | -2.3480281 | 1.2061752 | -1.9466725 | 0.0515740 | 0.0702767 |
¿Qué significa cada columna? Revisa la viñeta de DESeq2 aquí.
- Ahora establezcamos un umbral de significancia estadística para los valores de p-value ajustado o
padj. Cualquier resultado bajo este umbral será considerado no significativo y viceversa.
# Este es nuestro umbral
alpha = 0.01
# Ordenamos la tabla de resultados
res.joro.azul = res.joro.azul[order(res.joro.azul$padj, na.last=NA), ]
# Filtramos según nuestro umbral alpha
sigtab = res.joro.azul[(res.joro.azul$padj < alpha), ]
# Le agregamos la taxonomía a la tabla
sigtab = cbind(as(sigtab, "data.frame"), as(tax_table(psd5)[rownames(sigtab), ], "matrix"))
# Manipulaciones varias para finalmente graficar los resultados
sigtabgen = subset(sigtab, !is.na(Genus))
# Phylum order
x = tapply(sigtabgen$log2FoldChange, sigtabgen$Phylum, function(x) max(x))
x = sort(x, TRUE)
sigtabgen$Phylum = factor(as.character(sigtabgen$Phylum), levels=names(x))
# Genus order
x = tapply(sigtabgen$log2FoldChange, sigtabgen$Genus, function(x) max(x))
x = sort(x, TRUE)
sigtabgen$Genus = factor(as.character(sigtabgen$Genus), levels=names(x))
ggplot(sigtabgen, aes(y=Genus, x=log2FoldChange, color=Phylum)) +
geom_vline(xintercept = 0.0, color = "gray", size = 0.5) +
geom_point(size=4) +
theme(axis.text.x = element_text(angle = -90, hjust = 0, vjust=0.5, size = 10), axis.text.y = element_text(size = 13), legend.text = element_text(size = 13) )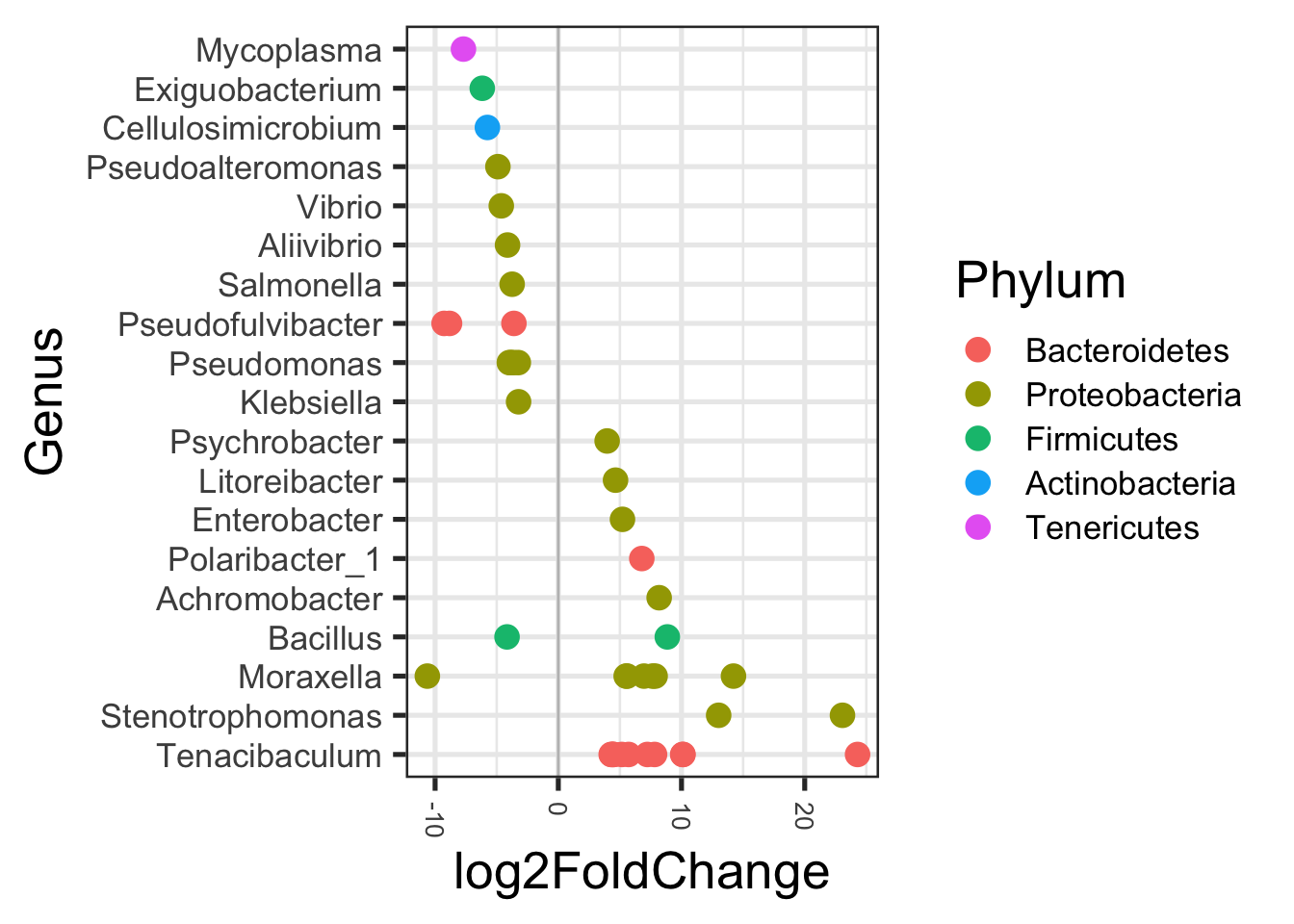
3.3.4 Redes de co-ocurrencia
Para finalizar, vamos a echar un vistazo a las capacidades de phyloseq para generar redes de co-occurrencia. Las redes de co-ocurrencia nos dan pistas sobre potenciales interacciones ecológicas entre organismos. Estas interacciones pueden ser directas o indirectas (no lo podemos determinar a partir de una red) y nos permiten comenzar a descifrar mecanismos ecológicos detrás de la composición de una comunidad microbiana. En general en ecología tenemos distintos tipos de interacciones:
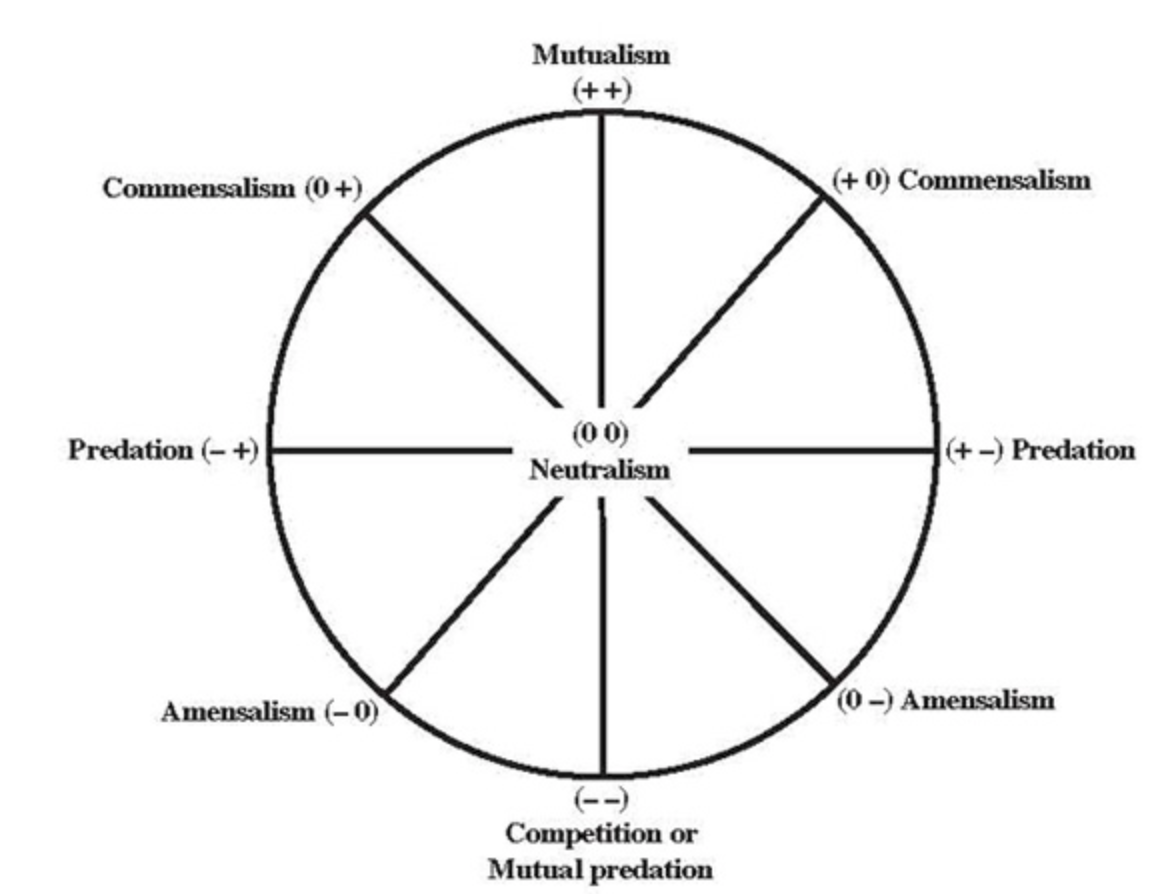
Donde destacan depredación, competición o depredación mutua, y mutualismo. Cada una de estas relaciones podría ser detectada en una red de co-ocurrencia según patrones de correlación positivos o negativos.
- Veamos como generaríamos una red en
phyloseq.
plot_net(psd5, type = "taxa", point_label = "Genus", point_size = 10, point_alpha = 0.5, maxdist = 0.5, color = "Phylum", distance = "bray", laymeth = "auto") 
La red generada con phyloseq no es una red de co-ocurrencia propiamente tal. Es más bien una red que representa relaciones de distancia entre taxa o muestras. En nuestro ejemplo usamos muestras. Para una red de co-ocurrencia propiamente tal necesitamos usar las funciones del paquete SpiecEasi.
- Veamos un ejemplo de cómo hacerlo.
se.mb.psd5 <- spiec.easi(psd5, method='mb', lambda.min.ratio=1e-2,
nlambda=20, icov.select.params=list(rep.num=50))
ig2.mb <- adj2igraph(se.mb.psd5$refit, vertex.attr=list(name=taxa_names(psd5)))
plot_network(ig2.mb, psd5, type='taxa', color="Phylum")
Para conocer más de redes de co-ocurrencia microbiana, dirígete al tutorial de la unidad Redes de co-ocurrencia de microorganismos